The Swern oxidation[1] is a class of “activated” dimethyl sulfoxide (DMSO) reaction in which the active species is a chlorodimethylsulfonium chloride salt. The mechanism of this transformation as shown in e.g. Wikipedia is illustrated below.‡ However, an interesting and important aspect of chemistry is not apparent in this schematic mechanism and to rectify this, a full computed mechanism is laid out below, for which the FAIR data has a DOI: 10.14469/hpc/13151


The first step involves attack of the oxygen of the DMSO on one carbon of the oxalyl chloride, which can be considered as an addition/elimination‡ substitution at the carbon. The departing chloride anion ends up loosely associated with the sulfur centre. The net result is that the trigonal bipyramidal sulfur is axially coordinated by the chlorine, but equatorially coordinated by the oxygen. The transition state for this step (TS1), shown at IRC = 0.0 in the above energy profile, has a relatively low activation barrier. Click on any animation to view 3D model.
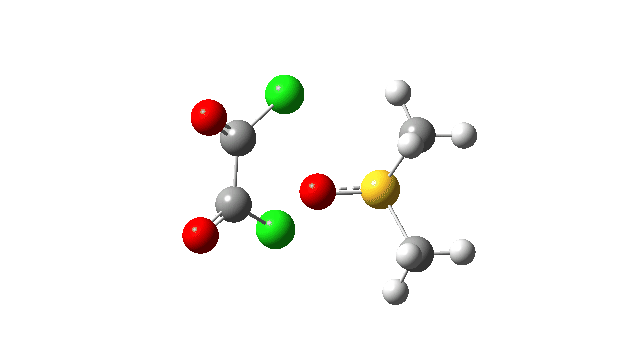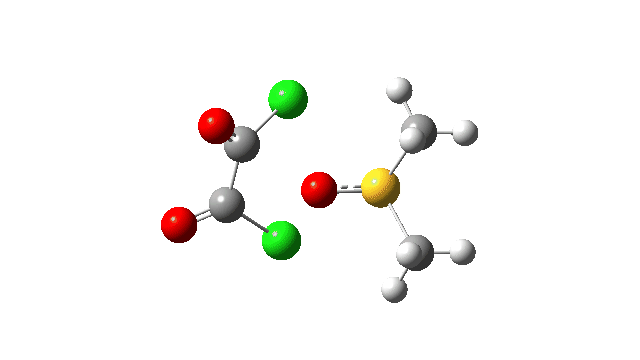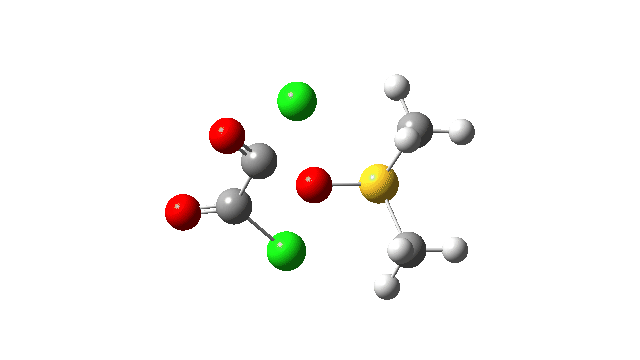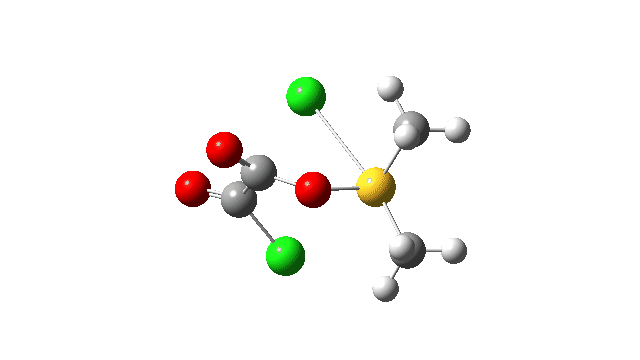
The key step is what is called a pseudorotation at the sulfur centre (TS2), which transforms the ax/eq relationship of the Cl/O atoms at the sulfur into an ax/ax one (TS at IRC +8.5 above). This is the energy high point along the reaction path. Note also the large increase in dipole moment, indicating ionic character, along the path involving TS1 and TS2.
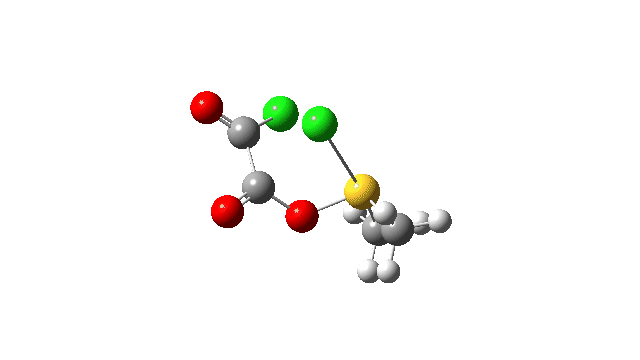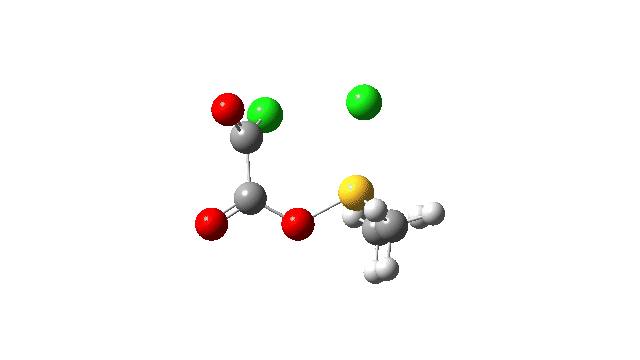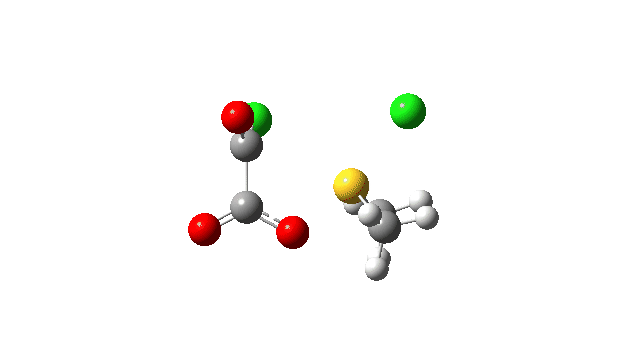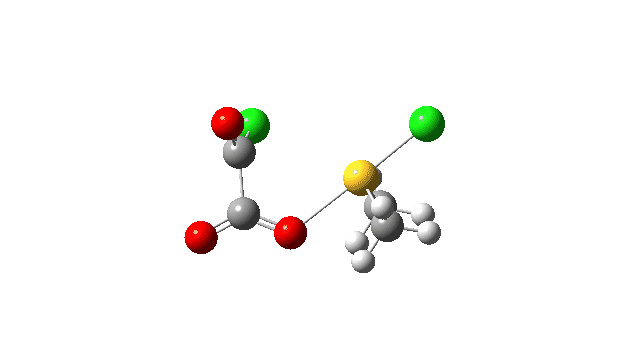

The S-O bond length response during this transformation is shown below. As the chlorine moves into this di-axial relationship, the S-O bond begins to weaken, from 1.635Å at the start, 1.675Å at the TS and 2.242Å at the end (Def2-TZVPP basis set).

This prepares the system for the final step (TS3), which is cleavage of the already weakened S-O bond (TS at IRC = 13.0 below, TS = 0.0 being the pseudorotation), accompanied by extrusion of CO, CO2 and Cl–. The liberated “ionic” chloride anion ends up loosely associated with the sulfur (2.88Å), whilst the “covalent” chlorine which had helped to evict the oxygen is 2.06Å.


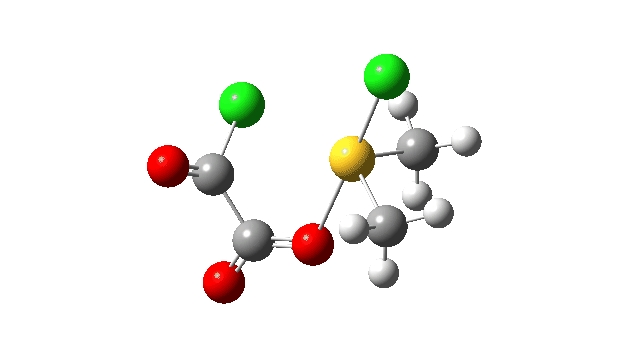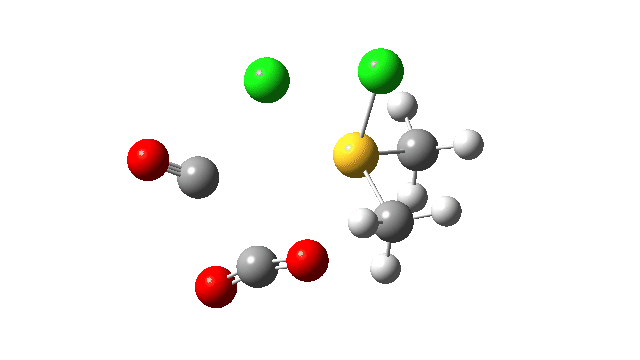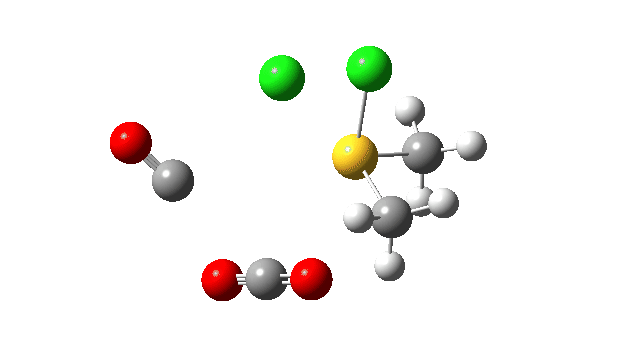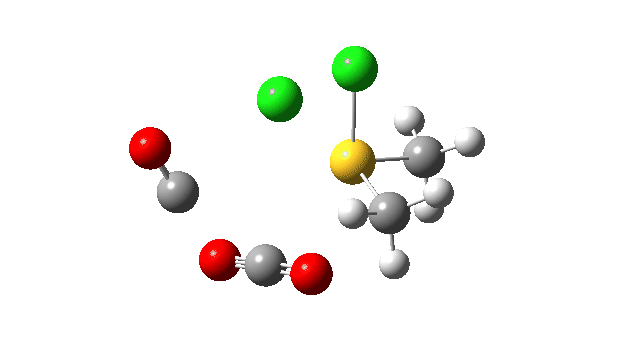
So to conclude, the mechanism of the formation of chlorodimethylsulfonium chloride is perhaps better illustrated as shown below involving the extra pseudorotation step, which as it happens is actually the rate determining step for this reaction. This pre-mechanism to the Swern oxidation is given little attention in most representations, such as the one at Wikipedia. But it actually contains a multitude of interesting (stereoelectronic) effects and is well worth teaching!

‡ Well, not quite. The Wiki version does not show the eliminating chloride anion in the first step (which is implied). The resulting curly arrows in the Wikipedia version are unbalanced and hence not formally correct! The double-headed arrow included in the representation above indicates an addition/elimination mechanism, which can be tracked by monitoring the carbonyl C=O bond length (@Def2-TZVPP). It starts at 1.181Å, reaches a maximum of 1.194Å just after the TS and then drops back to 1.186Å at the end as the chloride anion eliminates.

Citing this blog post: DOI 10.14469/hpc/13156
Author
References
- K. Omura, and D. Swern, "Oxidation of alcohols by “activated” dimethyl sulfoxide. a preparative, steric and mechanistic study", Tetrahedron, vol. 34, pp. 1651-1660, 1978. https://doi.org/10.1016/0040-4020(78)80197-5
I noticed that you performed the calculation for TS1 and TS2 with both s-cis and s-trans conformation in the oxalyl unit, but for TS3 only with the s-cis conformation.
Probably s-trans is preferred as it corresponds to the ground-state.
However, does the fragmentation also “work” (computationally) in the non-cyclic arrangement?
I assume the (pseudo)-cyclic TS3 gives a nicer (exergonic) enthalpy profile, since chloride ends up next to positive sulfur, so there is less charge separation?
(With “Probably s-trans is preferred” I only mean for T1 and T2, not T3).
Lukas,
The energies (b3lyp/empiricaldispersion=gd3bj/Def2-TZVPP) are as follows
1. Reactant (s-trans) -1700.503340 (s-cis), -1700.501382 (+1.2 kcal). There are significant interactions between the oxygen from DMSO and both carbonyl carbons and weaker ones between both chlorines and S in the DMSO in the s-cis reactant, (2.5A), which make for an interesting complex!
2. TS1 deriving from an s-cis rotamer of the reactant: -1700.483214
3. TS1 deriving from an s-trans rotamer of the reactant: -1700.487258 (-2.5 kcal/mol)
4. TS2 deriving from s-cis rotamer of TS1: -1700.476097
5. TS2 deriving from s-trans rotamer of TS1: -1700.477737 (-1.0)
6. TS3: -1700.501179.
7. Product: -1700.562974
So the s-cis pathway is slightly less stable than the s-trans. However, the product of TS2 (s-cis) aligns perfectly with the start point of TS3, as shown in the concatenated IRC plots. There is a very slight mis-match in energy between the end point of TS2 (s-trans) and the start point of TS3, due to a rotamer mis-match. So there might be a rotameric TS connecting TS2 and TS3, but likely of very low energy.
Re “I assume the (pseudo)-cyclic TS3 gives a nicer (exergonic) enthalpy profile, since chloride ends up next to positive sulfur, so there is less charge separation?”
Correct.