This is a venerable organic reaction, which curiously I have not previously covered here. First described in 1859, its nature was only properly elucidated in 1873. It is a member of a class of reaction I have previously named “solvolytically assisted pericyclic”, or “perisolvolytic“. Here I explore some of the subtle stereoelectronic effects observed for this apparently simple reaction.

It applies to a class of molecule known as 1,2-diols. Protonation is quickly followed by migration of a (in this example) methyl group, followed by deprotonation of the carbonyl group formed by this process. There are two mechanistic stages, the first being the departure of the now protonated “ol” unit, and the second the migration of the methyl. In most text books and of course Wikipedia, these are shown as very distinct steps. But they could also occur in one concerted step, albeit probably asynchronously.


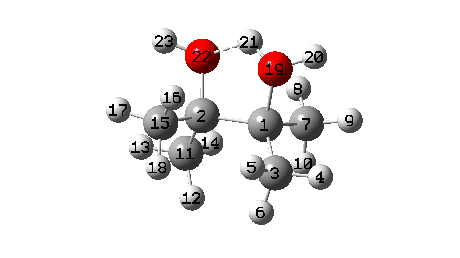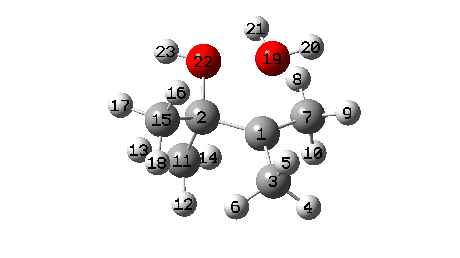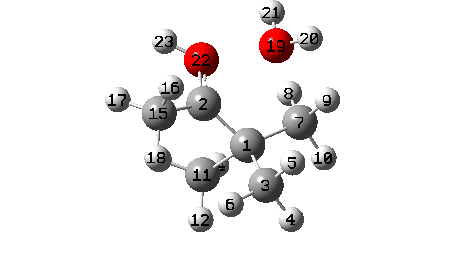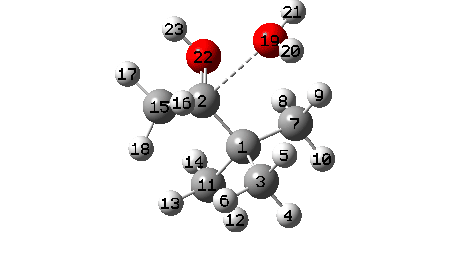
A B3LYP+GD3+BJ/Def2-TZVPP/SCRF=ethanol calculation provides mechanistic detail (FAIR Data 10.14469/hpc/1769)
- To start with, we note the H-bond formed between O22-H21. Between IRC = -10 and -6, this lengthens from 1.625Å to 1.843Å, destabilising the protonated alcohol group.
- Between IRC -6 to -1, the C1-O19 bond breaks, from a starting length of 1.556Å to ~2.787Å.
- When IRC 0.0 is reached (the transition state), the C11 methyl starts to migrate across, a process mostly complete by IRC +2.
- The final stage is formation of a weak interaction between C2 and O19 to reach IRC 7.
- Several more minor effects can also be discerned. Firstly methyl C3 rotates, to set up a better hyperconjugative interaction with the temporary carbocation forming at C1. This rotamer forms the first of several “hidden intermediates” in the reaction, intermediates which almost form before being consumed, at IRC -6.5 (see the plot above labelled RMS gradient form, for the minimum in the function at this IRC value).
- Another hidden intermediate appears at IRC -2, being the transient carbocation, as shown in stepwise versions of this mechanism, such as the Wikipedia page. But its not real, merely hidden! As it approaches, methyl C7 rotates to maximise the hyperconjugative interactions.
- At IRC ~+3, methyl C15 rotates to again maximise hyperconjugation with the newly formed C=O bond.
Ca we quantify some of these effects? This can be done by computing localised orbitals (NBOs) and pairwise interactions between a donor NBO (a bond or a lone pair) and an acceptor NBO (an antibonding orbital).
- The E(2) interaction between donor bond C2-C11 and acceptor C1-O19 is 3.3 kcal/mol (above the noise, but not especially strong). It corresponds to an antiperiplanar alignment of the C2-C11 σ orbital and the C1-O19 σ* orbitals and results in the breaking of bond C2-C11 (and reformation as C1-C11).
- The E(2) value between donor lone pair O22 and acceptor C2-C11 σ* is 6.9 kcal/mol and corresponds to antiperiplanar alignment of these two orbitals, resulting in formation of the C=O carbonyl π-bond, whilst simultaneously increasing the antibonding character of the C-C bond to encourage it to break.
Models of these two interactions can be seen below. Click on the image to load them. The colour blue overlaps positively with the colour purple, and red with orange.
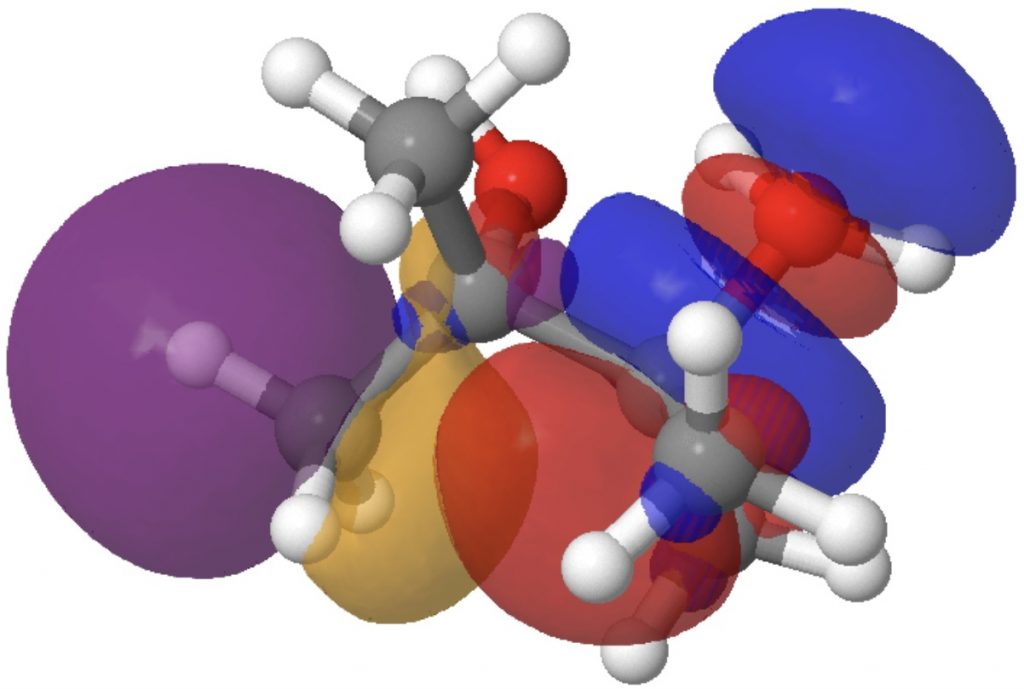
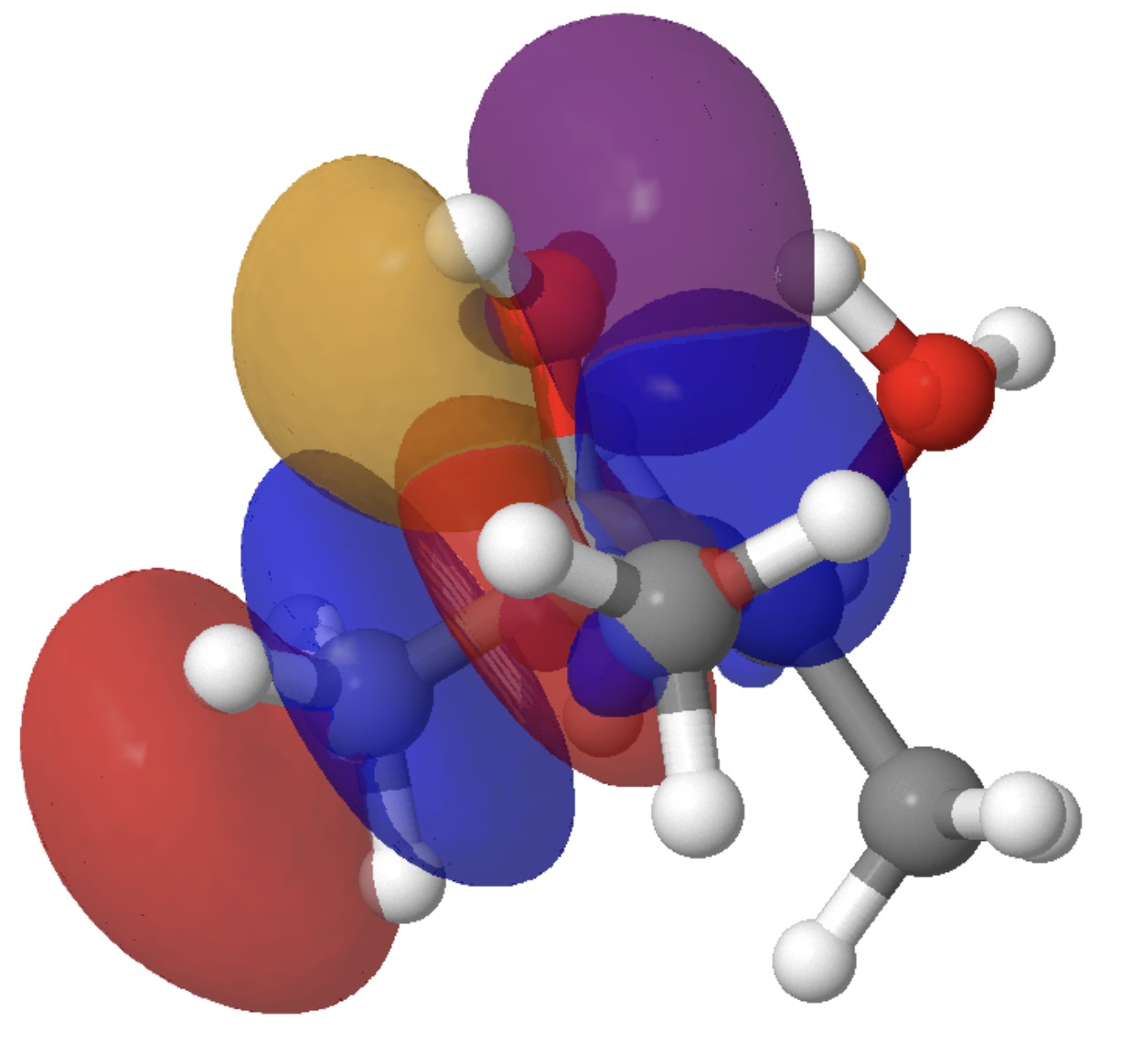
By the time the transition state is reached, these two interactions have evolved to the following:
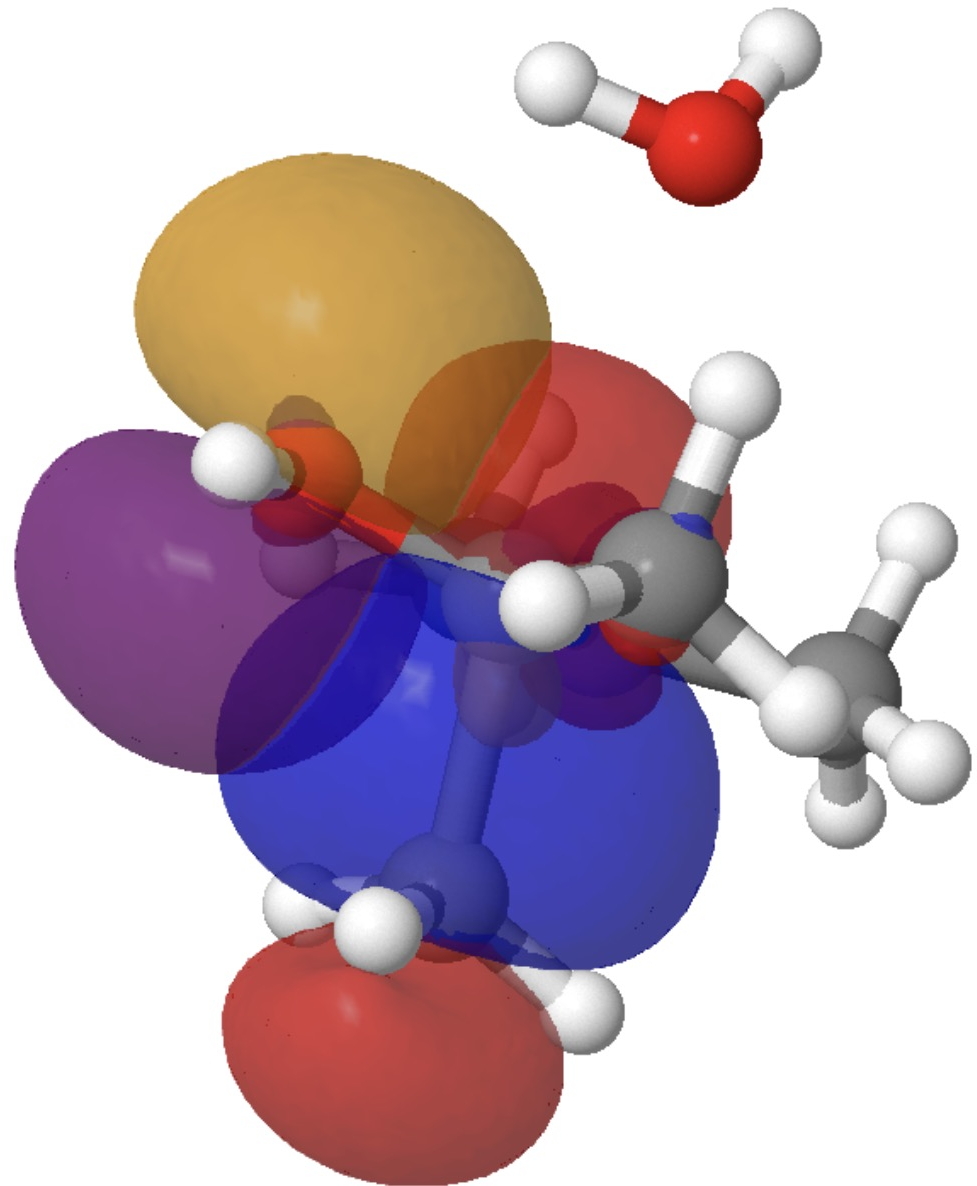
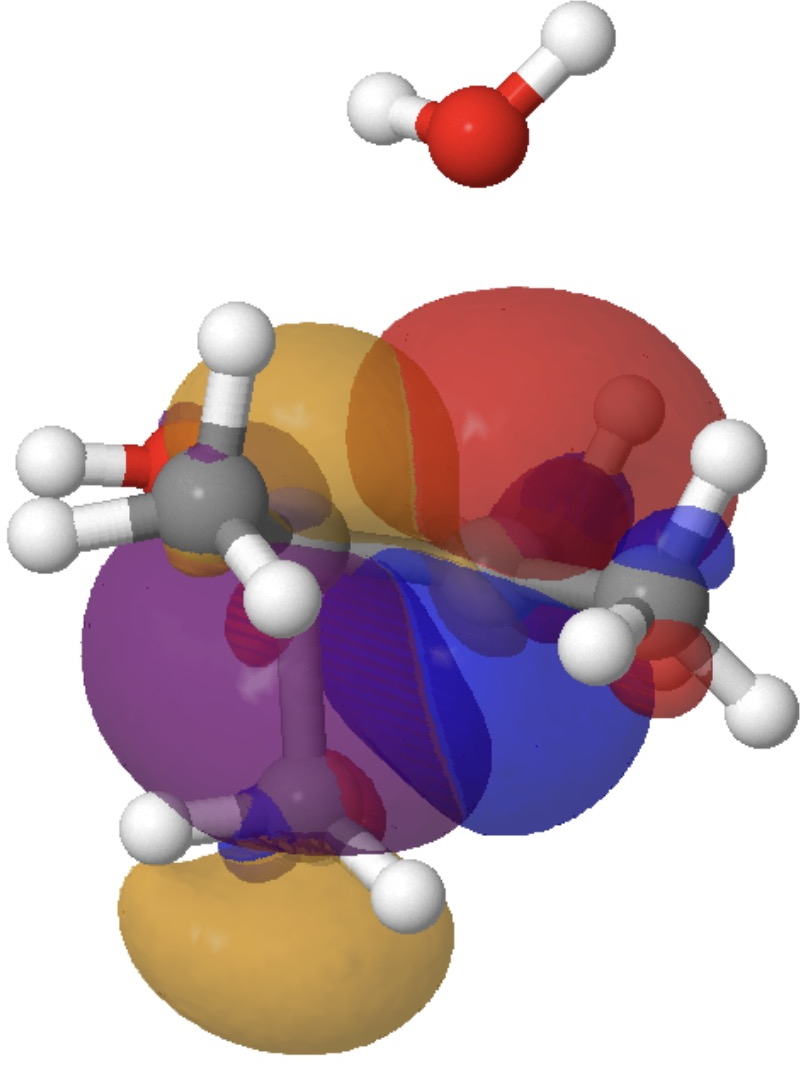
So this venerable reaction has some nice subtle stereoelectronic behaviour. Those methyl rotations have been skipped over here, but a deeper look into them might also be worthwhile. There is much more to this reaction, but I will leave this analysis here.
This post has DOI https://doi.org/10.14469/hpc/12684
“Asynchronously” for a “concerted” reaction. This is a word I shall use. It was some time ago, but if I recall correctly, Arigoni (sp?) argued there was no such thing as a concerted reaction. I have been an adherent of this concept as in if time can be divided to infinitely small intervals, then three balls cannot collide at the same time, two will always collide before the third.
I find this to be a useful construct beyond the pinacol rearrangement. I prefer to think of concerted and stepwise as the bounds of reactions, and actual reactions fall in between.
Strictly speaking, one should talk about trajectories and their behaviour. But in the absence of the statistics of trajectories, the next most useful interpretation is of the IRC (which is effectively just one, non statistical, trajectory). This charts the energy response between one minimum, through an energy maximum in one dimension, to another minimum. If this covers the reaction in question, then it is concerted. What is less commonly noticed is if the profile shows a “hidden intermediate”, defined loosely as a minimum in the gradient norm response rather than energy response. If that minimum is not zero, then the intermediate is regarded as “hidden”. In other words, its lifetime is only that of a molecular vibration and not longer. Such properties could be associated with one thing happening after another, ie asynchronous. These trajectories can be perturbed by many things. In this case, there seems little doubt that the presence of (heavy) counterions to the cation will also perturb the trajectories (as could solvent molecules). But getting the statistical distribution of trajectories for models that include counterions is non trivial.