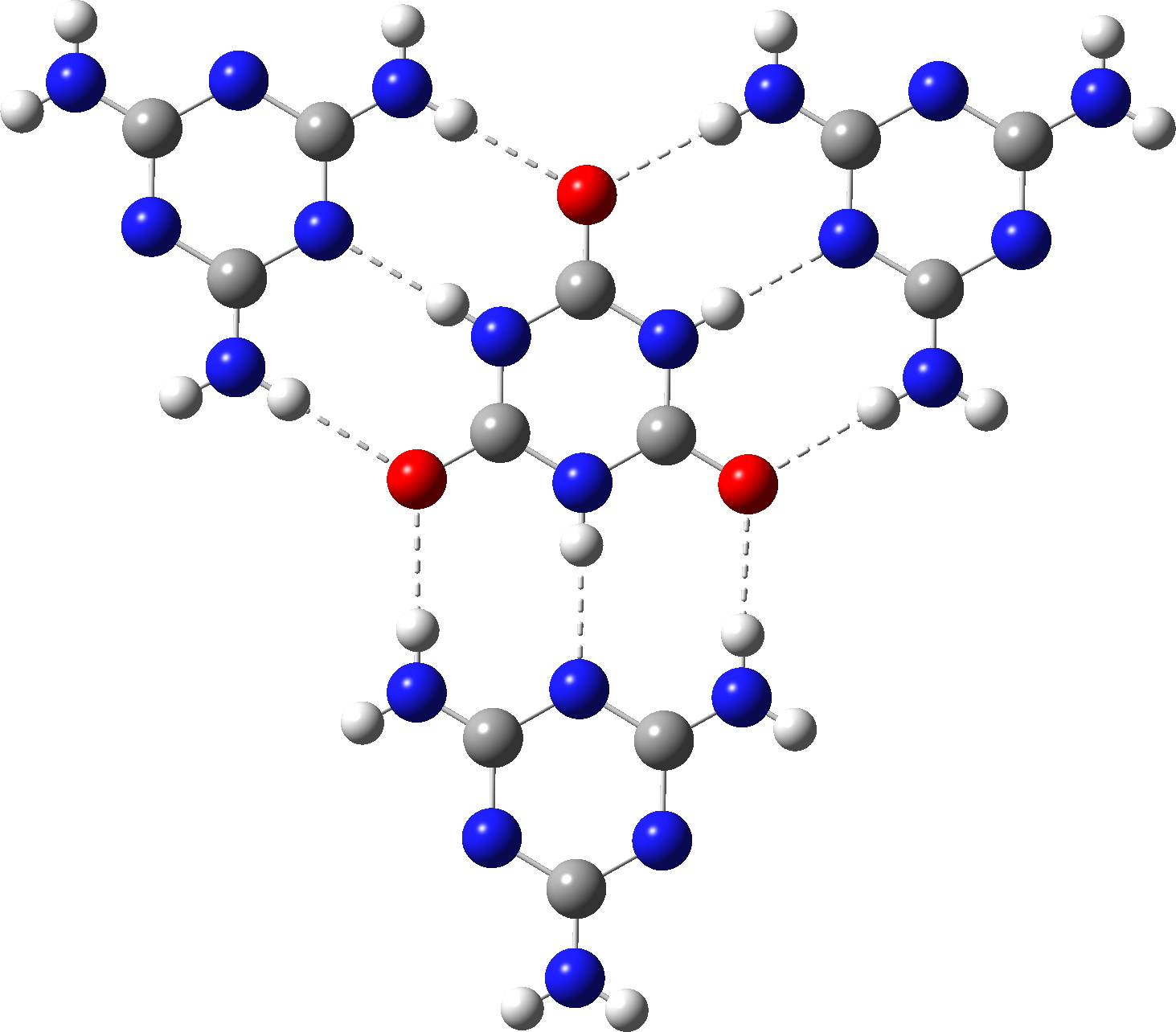
Some time ago in 2010, I showed a chemical problem I used to set during university entrance interviews. It was all about pattern recognition and how one can develop a hypothesis based on this. In that instance, it involved recognising that a cyclic molecule which appeared to have the cyclohexatriene benzene-aromatic pattern 1 was in fact a trimer of carbon dioxide. Perhaps small amounts of this aromatic molecule exist in solutions of fizzy drinks? Analysing these patterns occupied about 10-20 minutes of an interview, and although you might think I was posing a difficult challenge, many students successfully rose to it! Now I revisit, but with a slightly better reality check on a related molecule 2 (cyanuric acid).
. 
As many as 58 examples of crystal structures of 1,3,5-triazinane-2,4,6-trione 2 (cyanuric acid) are known, often with a co-adduct. Cyanuric acid is in effect a cyclic trimer of isocyanic acid rather than of carbon dioxide. These examples tend to be planar, with a mean C-N ring distance of ~1.37Å and a C-O distance of 1.22Å.
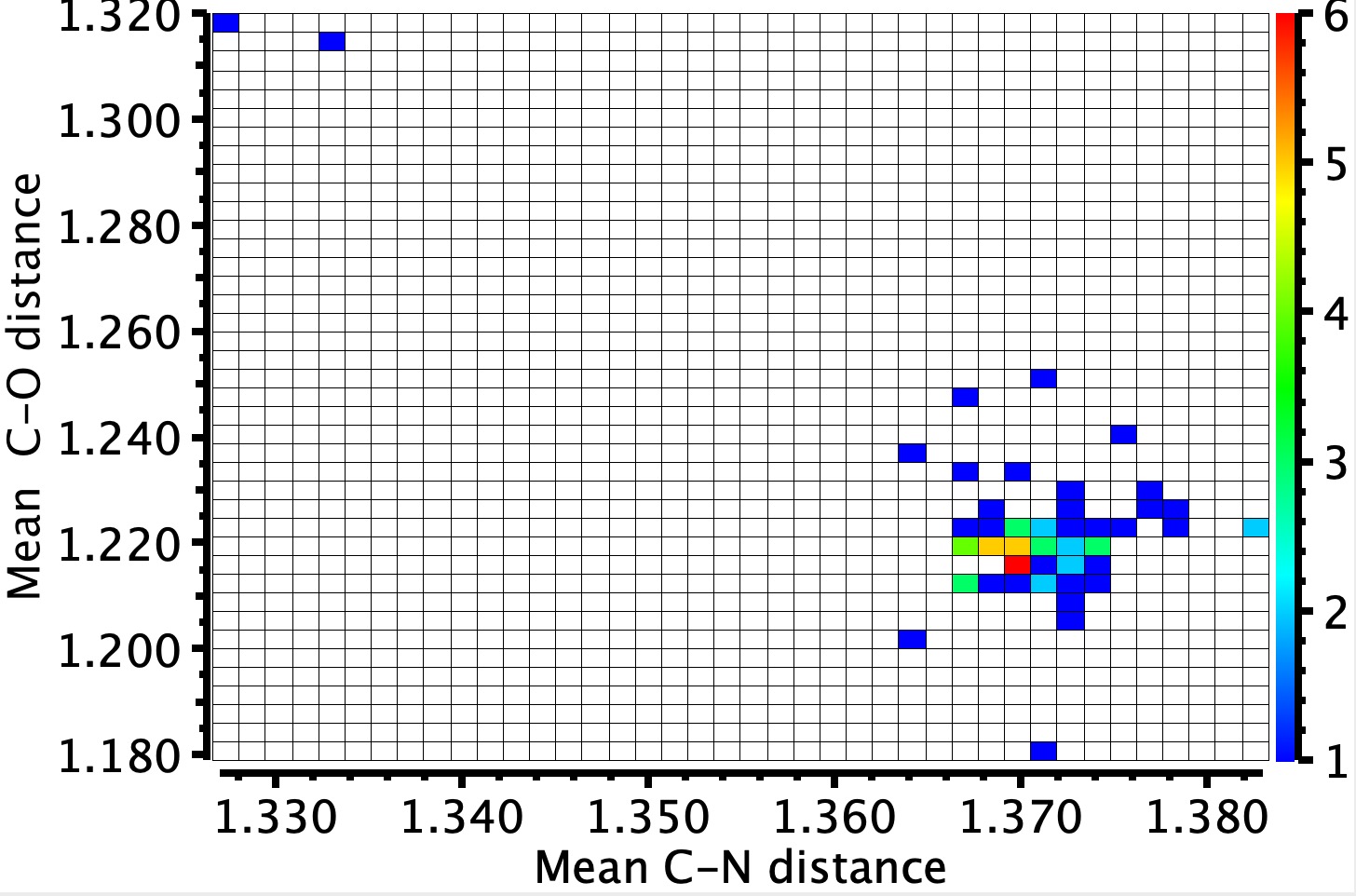
Two outliers stand out, both from a very recently published article, being a co-adduct with melamine (1,3,5-triazine-2,4,6-triamine).[1] QACSUI02 exhibits a shorter C-N distance of ~1.33Å but a longer C-O distances of 1.32Å and have a symmetrical patten of hydrogen bonds to the six receptors of the central unit. Could this correspond more closely to the cyclohexatriene resonance structures shown to the left of the diagram at the top? The first task is to see if these bond lengths can be replicated using calculation (often a useful procedure to check that the crystal structure is correct). For this purpose, the structure below was chosen as the starting point for various models, using an ωB97XD/Def2-TZVPP model.
| Model | C-N distance | C-O distance |
|---|---|---|
| QACSUI02 (crystal structure) | 1.331 | 1.318 |
| ωB97XD/Def2-TZVPP as single layer | 1.3678 | 1.2185 |
| ωB97XD/Def2-TZVPP three layers | 1.365 | 1.218 |
| ωB97XD/Def2-TZVPP no H-bonds | 1.3816 | 1.2002 |
|
|
||
| XAKSOU (crystal structure) | 1.367 | 1.208 |
| ωB97XD/Def2-TZVPP | 1.3670 | 1.2213 |
This creates a mystery. The calculated bond lengths show that whilst H-bonding to the central ring decreases the C-N length by 0.014Å and increases the C-O length by 0.017Å, this effect is nowhere near large enough to match the apparent lengths in the crystal structure, where a C-N effect of ~0.037Å would be needed.
Another system XAKSOU has been reported where discrete LiCl units replace the hydrogen the H-bonds formed to melamine above.[2] A Li is coordinated to the carbonyl oxygen instead of a hydrogen bond, and a chloride anion from another molecule in the unit cell replaces the H-bond to nitrogen.
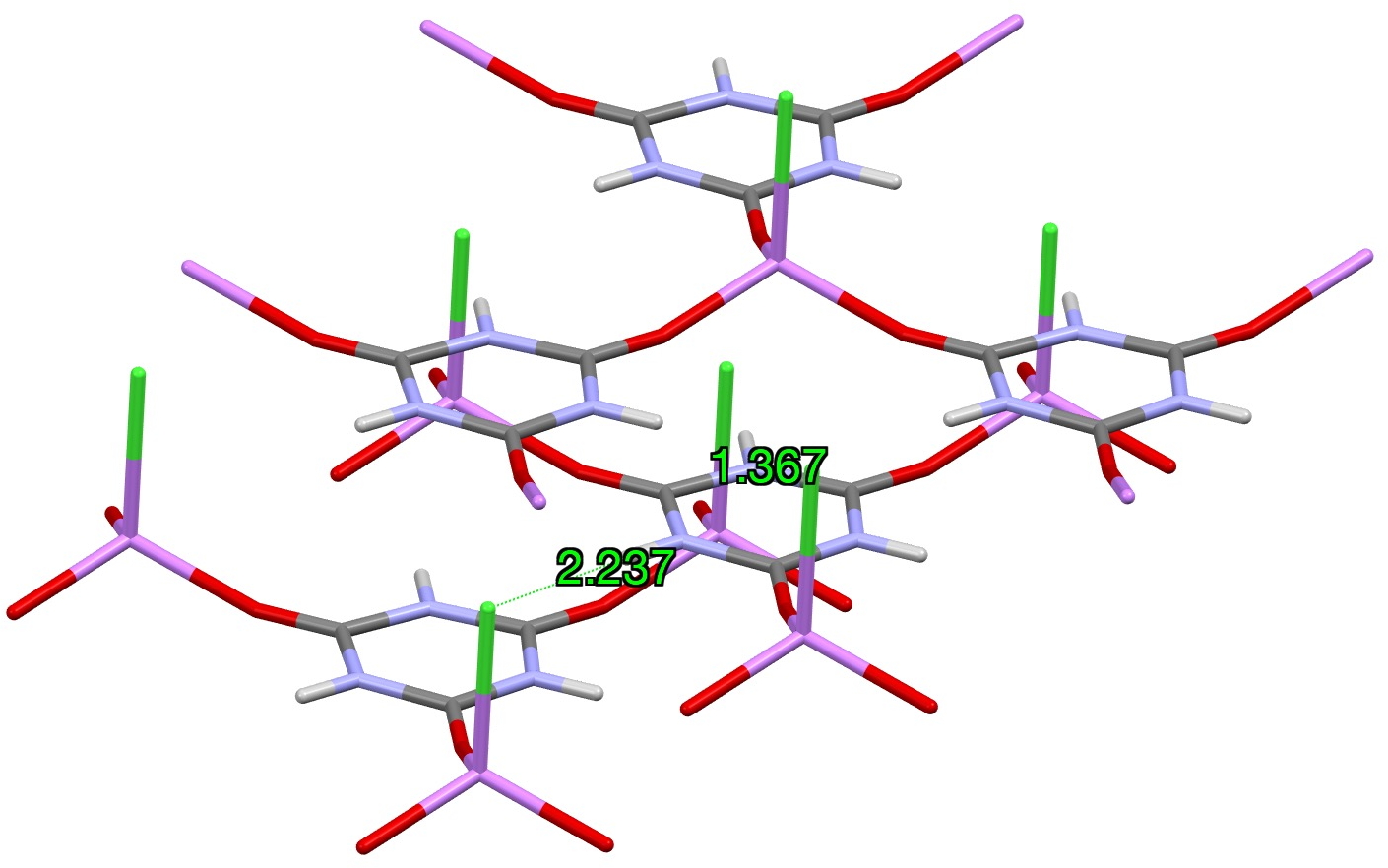
In the computed model, an intramolecular Cl-H hydrogen bond is used as the model, resulting in similar C-N lengths as the crystal structure (one which does not match the lengths in the outlying crystal structure QACSUI02)
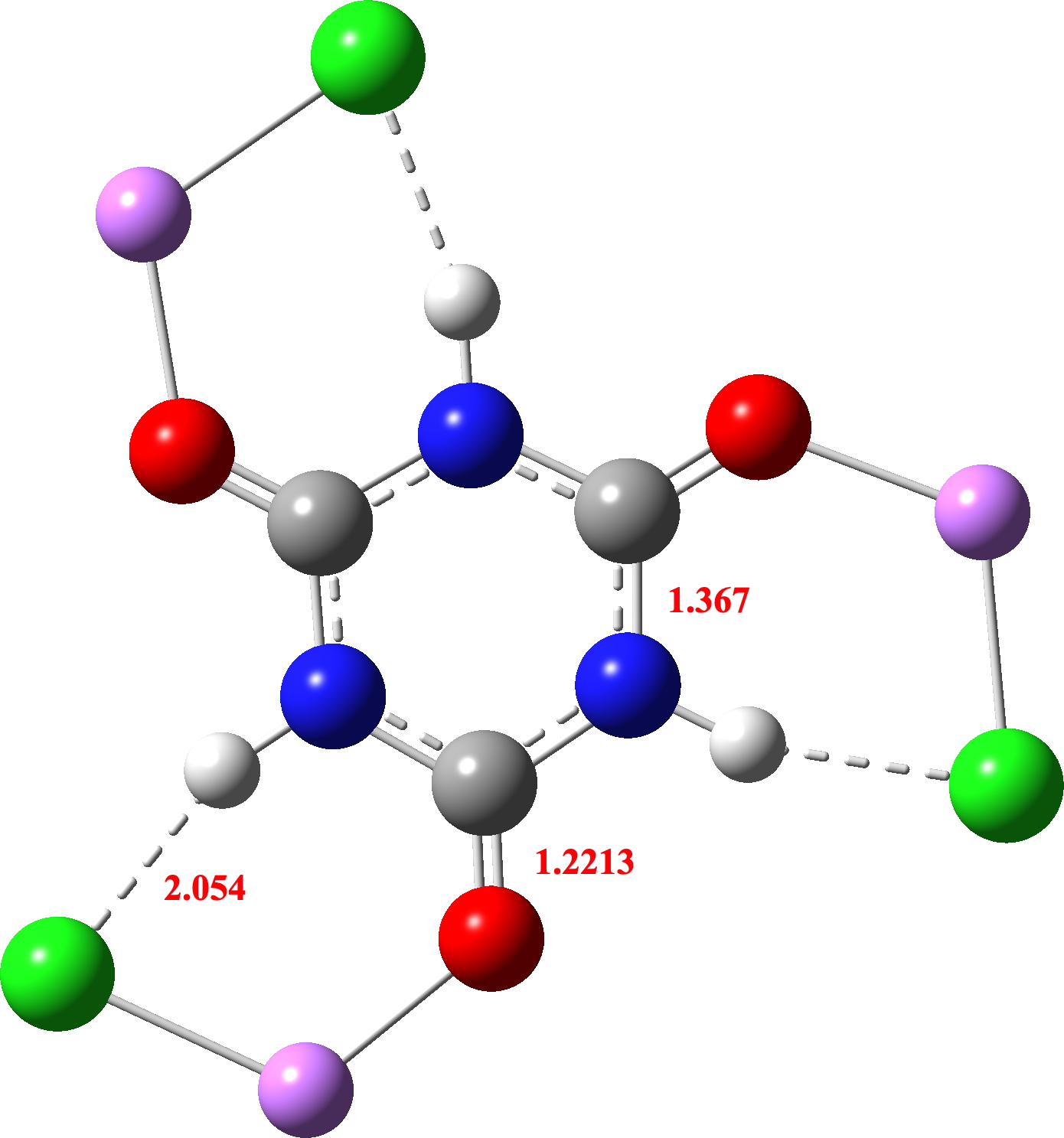
So the final question to ask is whether this latter structure is aromatic. NICS(0)/(1) values of -2.8/-1.1ppm are computed, which suggests very little aromaticity (aromatic values would be -10 to -20 pm). So it does not seem as if aromaticity can be tuned into cyanuric acid 2 by polarising both the NH and CO units with ionic/H-bond interactions so that the aromatic cyclohexatriene motif is better favoured over the 1,3,5-triazinane-2,4,6-trione non-aromatic resonance form. Are there any other examples where aromatically tunable molecules might be possible?
Author
References
- K. Song, H. Yang, B. Chen, X. Lin, Y. Liu, Y. Liu, H. Li, S. Zheng, and Z. Chen, "The facile implementing ternary resistive memory in graphite-like melamine-cyanuric acid hydrogen-bonded organic framework with high ternary yield and environmental tolerance", Applied Surface Science, vol. 608, pp. 155161, 2023. https://doi.org/10.1016/j.apsusc.2022.155161
- O. Shemchuk, D. Braga, L. Maini, and F. Grepioni, "Anhydrous ionic co-crystals of cyanuric acid with LiCl and NaCl", CrystEngComm, vol. 19, pp. 1366-1369, 2017. https://doi.org/10.1039/c7ce00037e
Hi Henry, I point you to the fantastic body of work by Judy Wu at Houston (http://jiwu.chem.uh.edu/) on this issue…