The last post addressed the concept of “steric clashes” in a pericyclic reaction transition state as an extension of the time honoured practice of building molecular models to analyse reaction outcomes. A modern computer generated model might express this in terms of a NCI (non-covalent-interaction) surface. A few posts ago, I had looked at some “molecules of the year” for 2020, one of which was a “figure-eight” twisted dodecaporphyrin in which an aspect of the reported[1] geometry had struck me as potentially lacking features due to the so-called non-covalent dispersion or van der Waals attractions. So I am revisiting here by adding the NCI surface for this molecule and one other.
The molecule in question has 720 atoms and can be regarded as a [162]-annulene (4n+2, n=40) with a linking number Lk =2π.[2] The NCI surface is ideally computed from a “self-consistent-field or SCF density” and so in this instance I used the PM7 SCF-density, which is derived from the valence shell only and does not include the core shells. That hardly matters since the non-covalent NCI surface does not use the core shells!
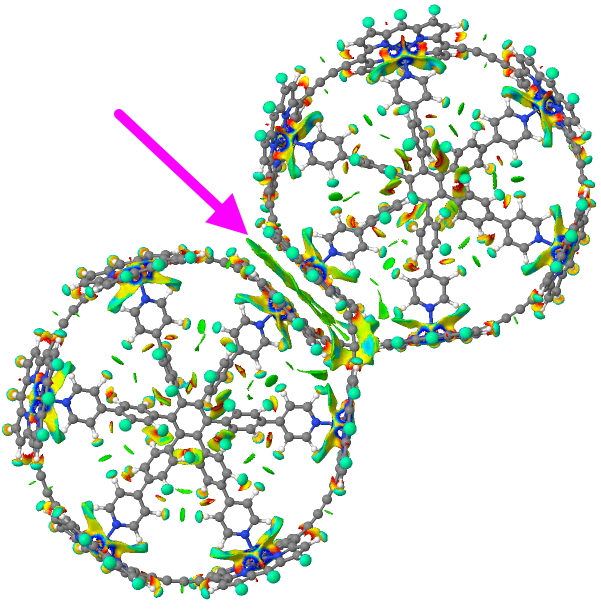
Click for 3D model
You can see from the above that the porphyrin-stacking region has a very dense NCI green surface (arrow), indicating a lot of stabilisation is originating there; something lacking in the original proposed structure. There are lots of other features and so I do encourage you to explore the 3D model.
The second (hypothetical) molecule is a simpler CH-based [144]-annulene,[3] comprising a twisted coil of 144 CH= units with a linking number Lk = 18π (the largest such ever proposed for a molecule!). The SCF-NCI surface (derived from an ωB97XD/6-31G(d,p) calculation) is contiguous all the way around the circuit and must be the ultimate π-π stacked molecule!
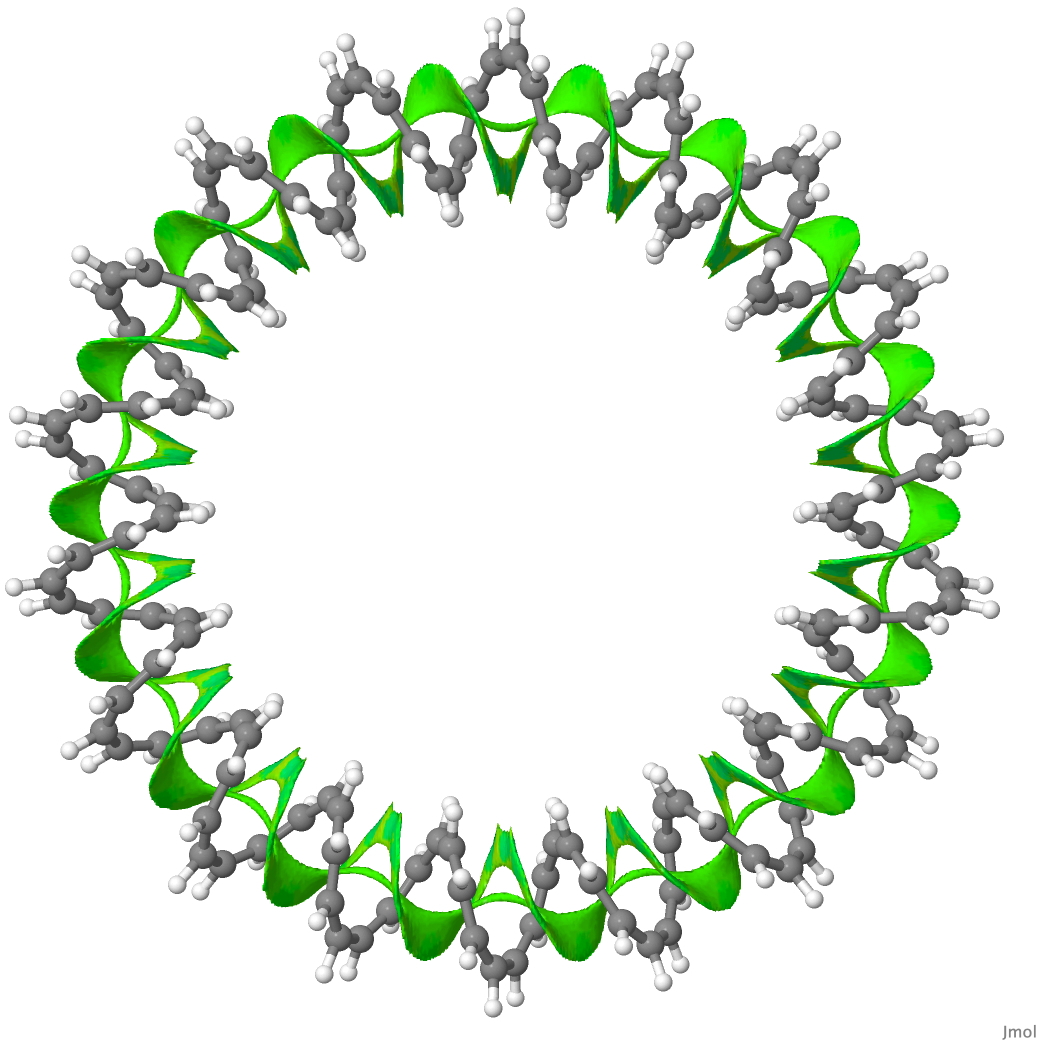
144-annulene. Click for 3D
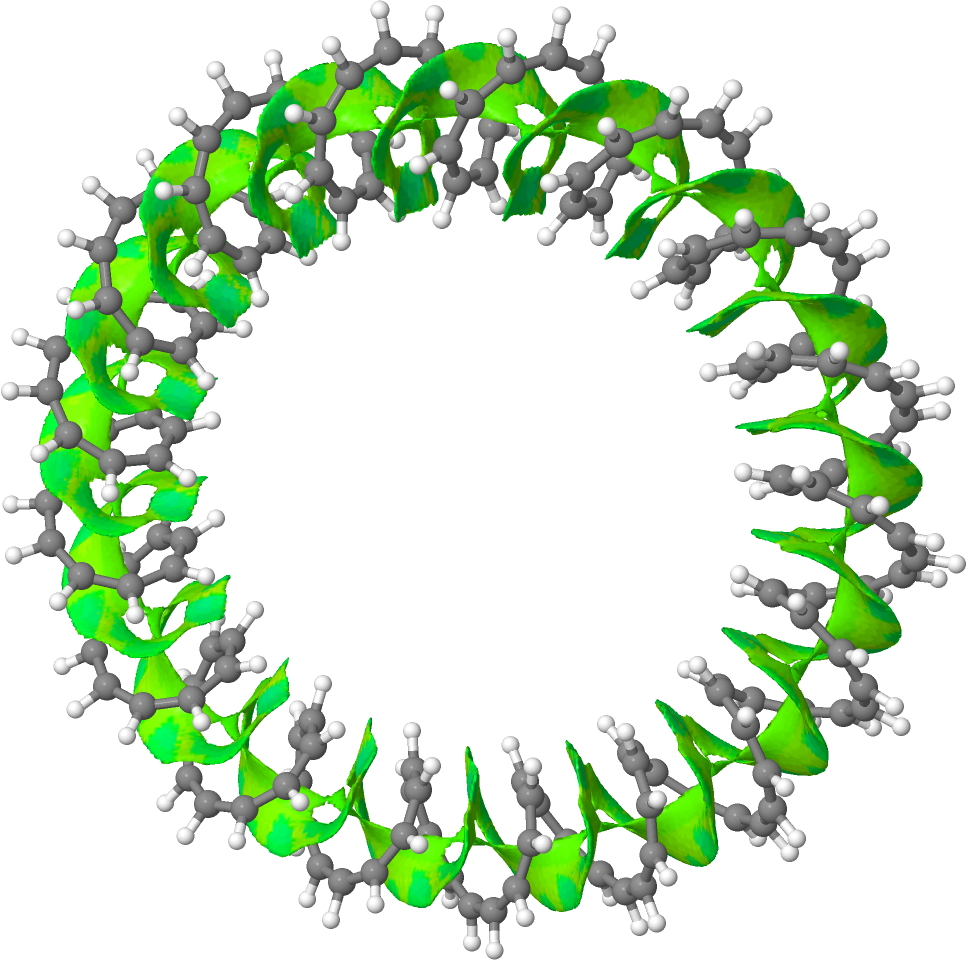
153-annulene. Click for 3D
I should end with a brief tutorial on how to generate these surfaces. You need a density matrix (e.g. DOI: 10.14469/ch/16967). In programs such as Gaussian 16, this can be obtained from the checkpoint file, which contains it. A progam called cubgen is used by (e.g.) Gaussian to create a 3D cube of electron density values (as well as other interesting properties). To get good resolution (~ 0.044Å) the file will be between 500 – 800 Mbyte in size. If a resolution of ~ 0.088Å is used it will be eight times smaller. For cube files less than ~105 Mbyte in size, you can use this Web-based tool (DOI: 10.14469/hpc/7864) to get the NCI surface. For the larger files you will need the Jmol application which can sustain files up to ~1 Gbyte (or larger, but I have not tested) and where you will run the following script:
load density.cub;isosurface parameters [0.5 1 0.0005 0.05 0.95 1.00] NCI "";color isosurface "bgyor" range -0.04 0.04;write density.xyz;write density.jvxl;
(where this script assumes that the file density.cub file is in the same folder as the Jmol.java application).
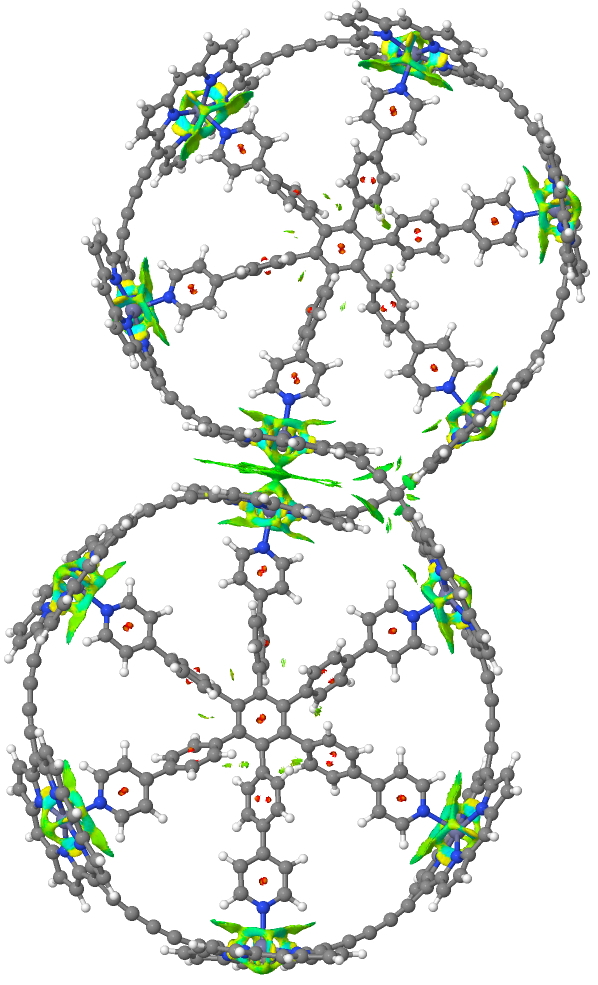
Postscript: The NCI analysis is based on computing the total density of the molecule. Close inspection of the top molecule as computed using the semi-empirical method PM7 reveals some interesting features extending beyond the C-H bonds. Analysis of this reveals it to be an artefact of the computed density, itself traced back to differences in how overlaps are handled in computing the density for this particular method. This error is not present for the MNDO semi-empirical method. When evaluated using MNDO, but at the geometry computed by PM7, these artefacts are removed. The NCI feature in the π-π stacking shown above however remains and hence is not an artefact.
Author
References
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M.D. Peeks, R. Haver, H. Jiang, T.D.W. Claridge, and H.L. Anderson, "Global aromaticity at the nanoscale", Nature Chemistry, vol. 12, pp. 236-241, 2020. https://doi.org/10.1038/s41557-019-0398-3
- S.M. Rappaport, and H.S. Rzepa, "Intrinsically Chiral Aromaticity. Rules Incorporating Linking Number, Twist, and Writhe for Higher-Twist Möbius Annulenes", Journal of the American Chemical Society, vol. 130, pp. 7613-7619, 2008. https://doi.org/10.1021/ja710438j
- R.J.F. Berger, "Prediction of a Cyclic Helical Oligoacetylene Showing Anapolar Ring Currents in the Magnetic Field", Zeitschrift für Naturforschung B, vol. 67, pp. 1127-1131, 2012. https://doi.org/10.5560/znb.2012-0189