The quote of the post title comes from R. B. Woodward explaining the genesis of the discovery of what are now known as the Woodward-Hoffmann rules for pericyclic reactions.[1] I first wrote about this in 2012, noting that “for (that) blog, I do not want to investigate the transition states”. Here I take a closer look at this aspect.

I will start by explaining my then reluctance to discuss transition states. Woodward in describing this discovery (in Chem. Soc. Special Publications (Aromaticity), 1967, 21, 217; a historic article which unfortunately remains off-line) notes the “steric preference for attack below the plane for C-5 and a gentle spiral for the cyclization to achieve the required stereochemistry at C-6″. In reference to the diagram above, he is talking about the reaction G to J which he thought was favoured over G to H on steric grounds. We must now try to judge what criteria might have been used to establish these steric grounds. He might have been referring to the relative thermodynamic stabilities of H vs J, which is the aspect I addressed in my earlier blog. But it has now been pointed out to me‡ that Woodward is more likely to have been thinking about the transition state for the reaction, in referring to a “gentle spiral” for the reaction path as inferred by model building. So why my reluctance in 2012 to look at this aspect? As Woodward himself quickly came to realise, the transition state for G to H is electronically “allowed” but the transition state for G to J is electronically “forbidden”. Let me qualify that. The latter is only forbidden on the ground state electronic surface, but it is allowed on an open shell excited state (photochemical) surface. It is very difficult (if not impossible) to directly compare the energies of these two electronic states for any steric differences that might be hidden or embedded within them. So how did Woodward initially infer a “steric preference” between these two reactions?
Model building reached its peak as an essential tool for understanding chemistry in the 1950s, with the likes of Pauling and Watson + Crick making Nobel-prize winning discoveries using this technique. By the 1960s, one could buy commercial model building kits, such as Dreiding stereomodels (1958) which focused on the bonds themselves and CPK or spacefilling models (~1952[2]) based on the size of the atom (a technique pioneered by Loschmidt as long ago as 1860). I would point out that such models are constructed for molecules in their presumed ground electronic state! So Woodward must have been constructing models for G to H and G to J with the implicit assumption that they were in the ground electronic state. Clearly he noticed something which led him to conclude that these models predicted G to J over G to H. I do not know if his models have survived to posterity and are now in a museum somewhere; the chances are we will never know exactly what it was that alerted him that the formation of G to H was so unexpected that it triggered a Nobel-prize winning theory!
Having declined to build TS models in my original musings on this topic, I now decided to bite the bullet and try to now locate at least approximate models for both possible stereochemical outcomes. The disrotatory transition state for G to H is relatively trivial. Here I used the PM7 method, which I noted previously nicely absorbs dispersion corrections which may be important! It also allows a full IRC for the reaction path to be constructed in just a few hours (a DFT approach would take quite a lot longer). The FAIR data for my models can be found at DOI: 10.14469/hpc/7806

I then realised that the electronically “forbidden” transformation G to J (something that makes locating a transition state on the ground state surface unlikely) was in fact allowed for an open shell triplet state (a excited state). In this state, transition state location actually proceeds without issue to find a nice conrotatory transition state.

The two key transition state models are each shown below in two representations. The clashes noted are approaches of two atoms closer than the sum of the van der Waals radii. First, I note that transition state G to H clashes a hydrogen with the adjacent methyl group (H…H contact 1.937Å using the PM7 semi-empirical method, 1.942Å using the ωB97XD/6-311G(d,p) density functional method).†
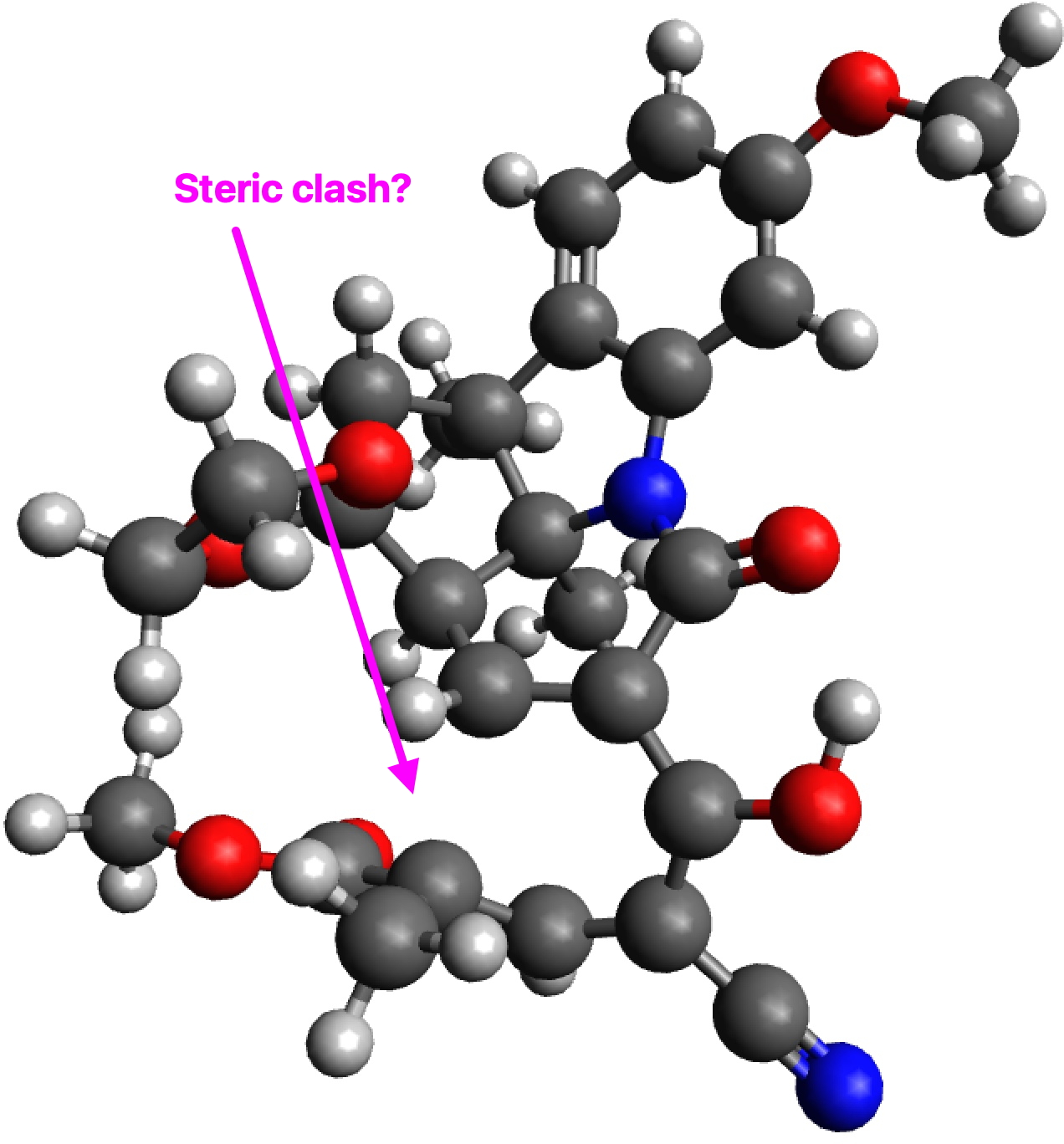
G to H, ball and stick representation. Click to view 3D♥
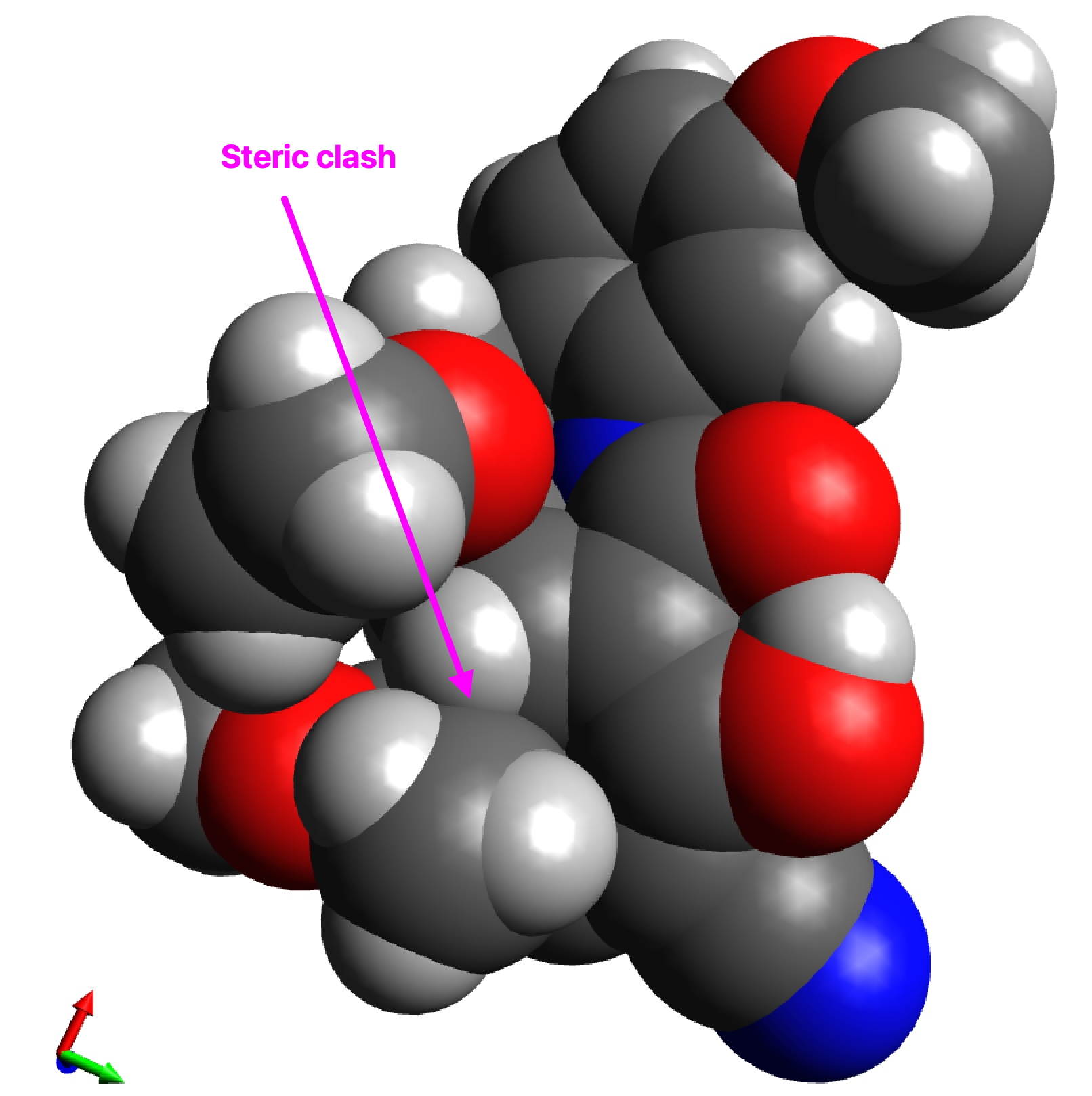
G to H, spacefilling representation
G to J also exhibits a clash, albeit a lesser one, between the hydrogens of two methyl groups (2.01Å for PM7, 2.03Å for ωB97XD/6-311G(d,p)). So one could argue that G to J is indeed favoured on steric grounds over G to H, but only by about 0.07Å in the close approach of pairs of non-bonded hydrogen atoms. I also note that Woodward’s gentle spiral or spiral of low pitch is in fact a left-handed one!
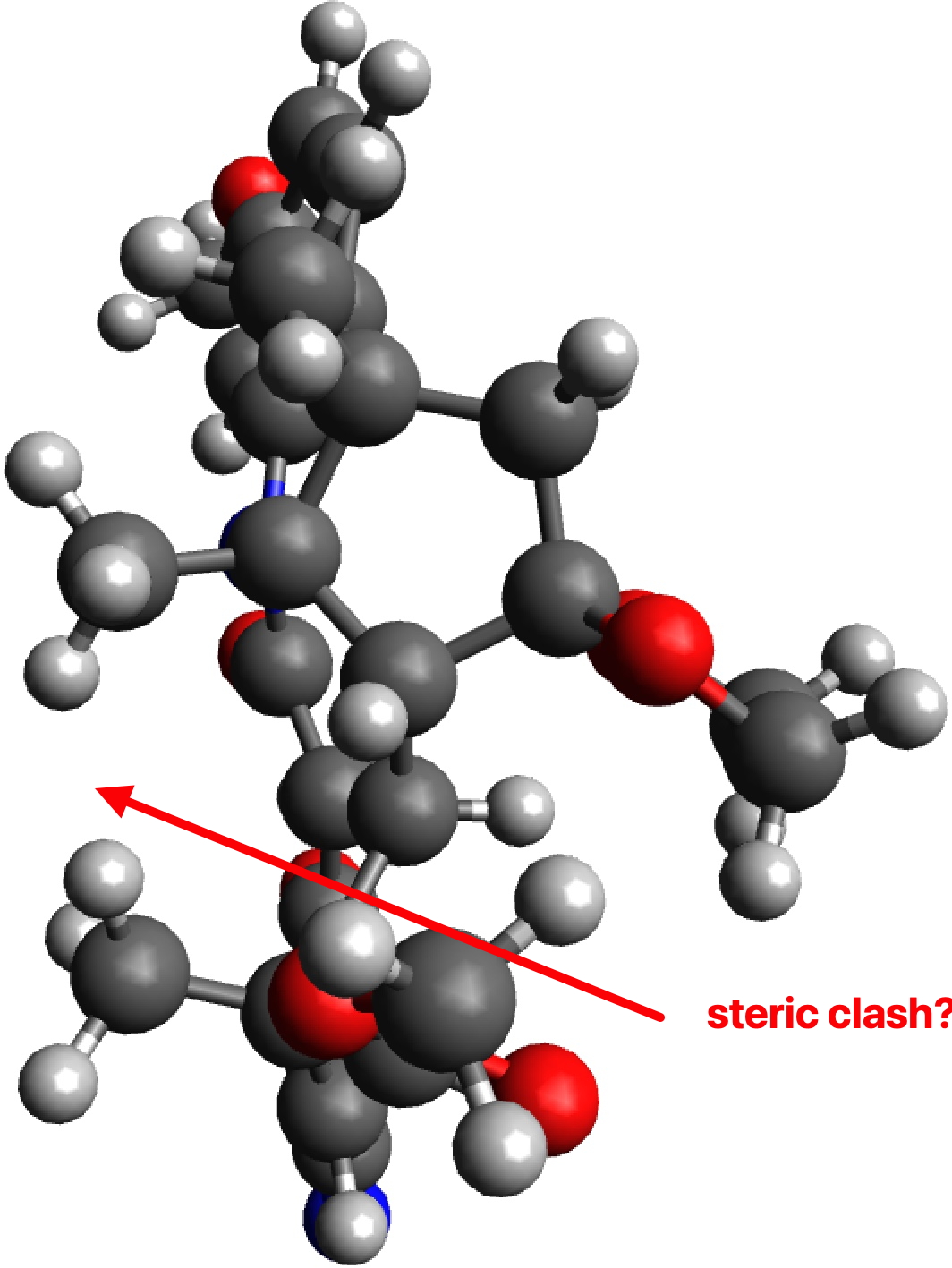
G to J, ball and stick representation. Click to view 3D
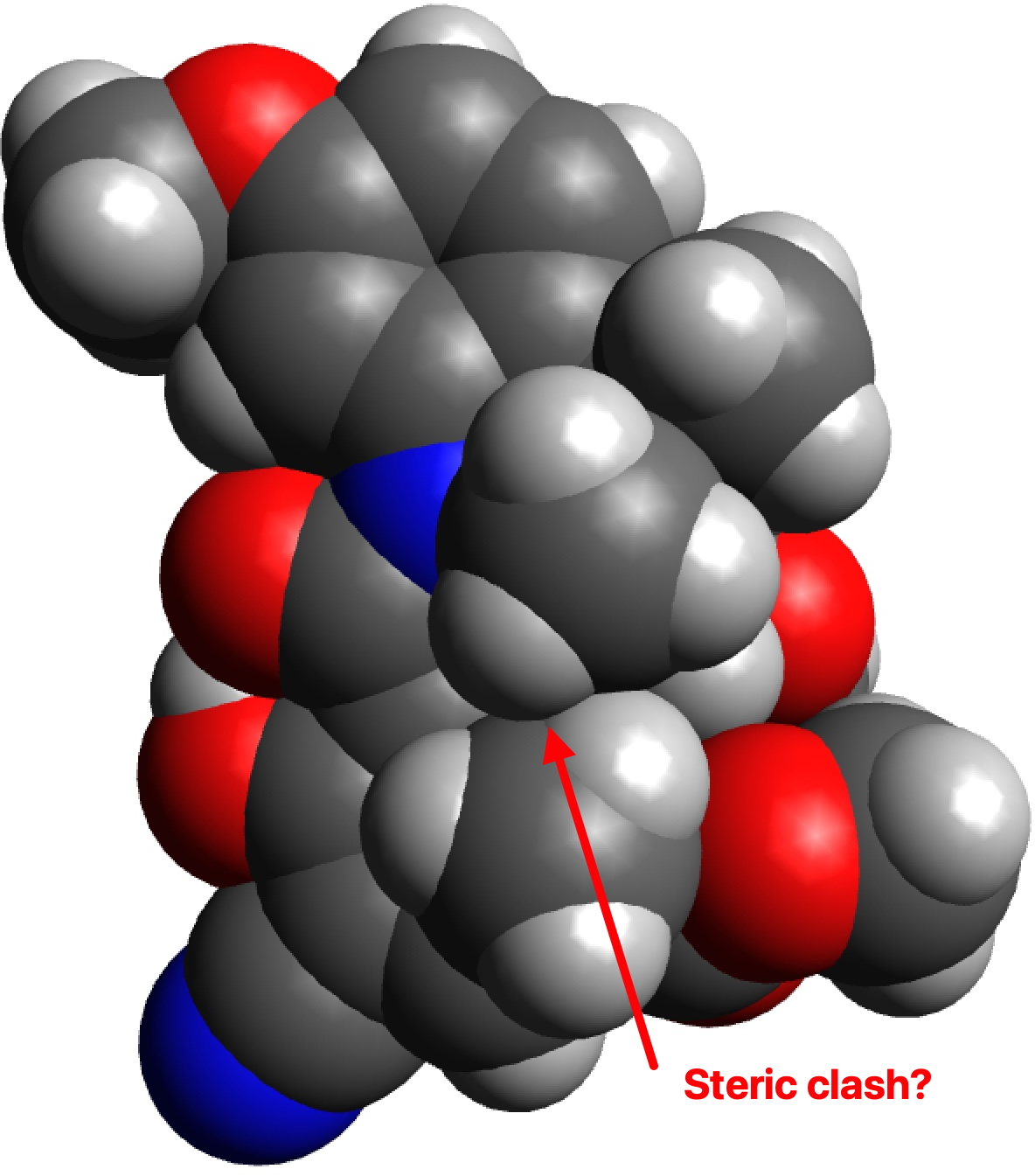
G to J, spacefilling representation.
To get another perspective on what this means in reality, I conducted a search of the CSD (Cambridge structure database) for the sub-structure shown below:
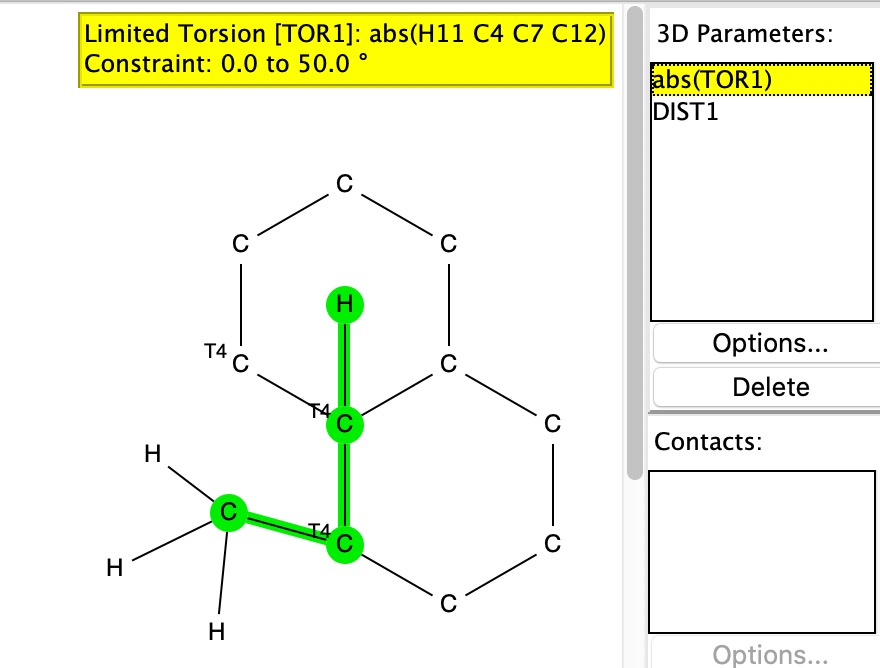
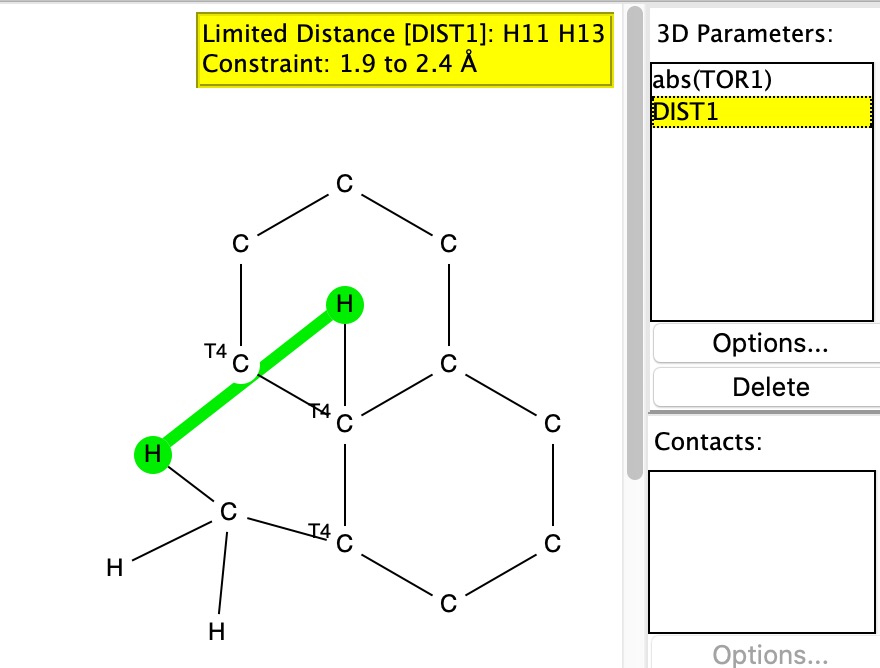
The results show H…H contacts down to about 2.03Å, which suggests that the steric clash for G to H probably is slightly repulsive, whilst that for G to J could be on the verge of being attractive.
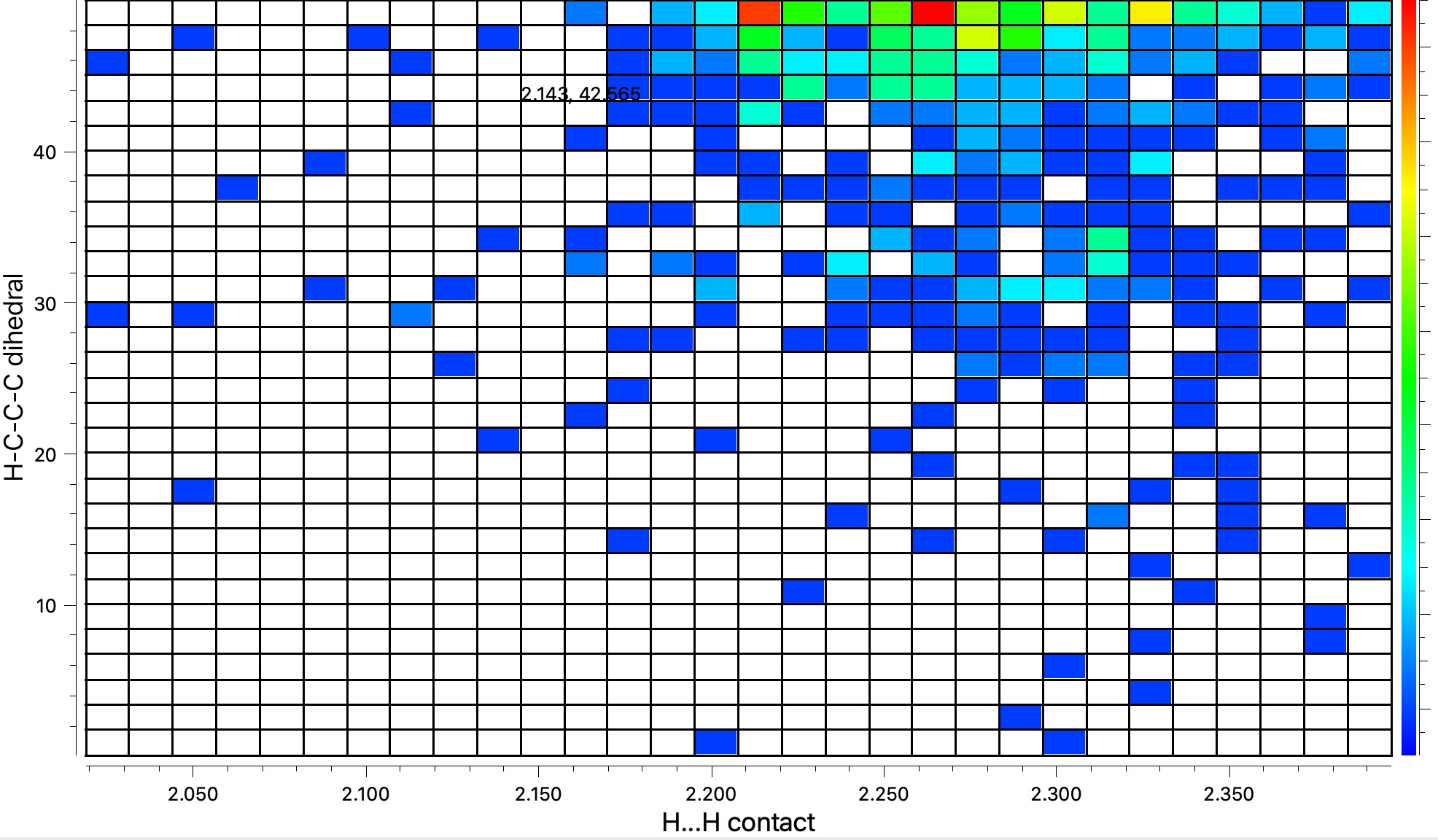
We might conclude that there is probably only a small steric difference between the two quantitative reaction models G to H and G to J as evaluated here, probably favouring the latter and assuming that the sterics are expressed entirely by van der Waals distances and have not been absorbed into bond angles etc. Of course much of what I have done and explained here was not common in the 1960s. The details of how Woodward’s models were actually constructed and how quantitative they were may never be discovered. It matters not of course, since the surprise of finding the actual product was H and not J went on to catalyse one of the great theories of organic chemistry!
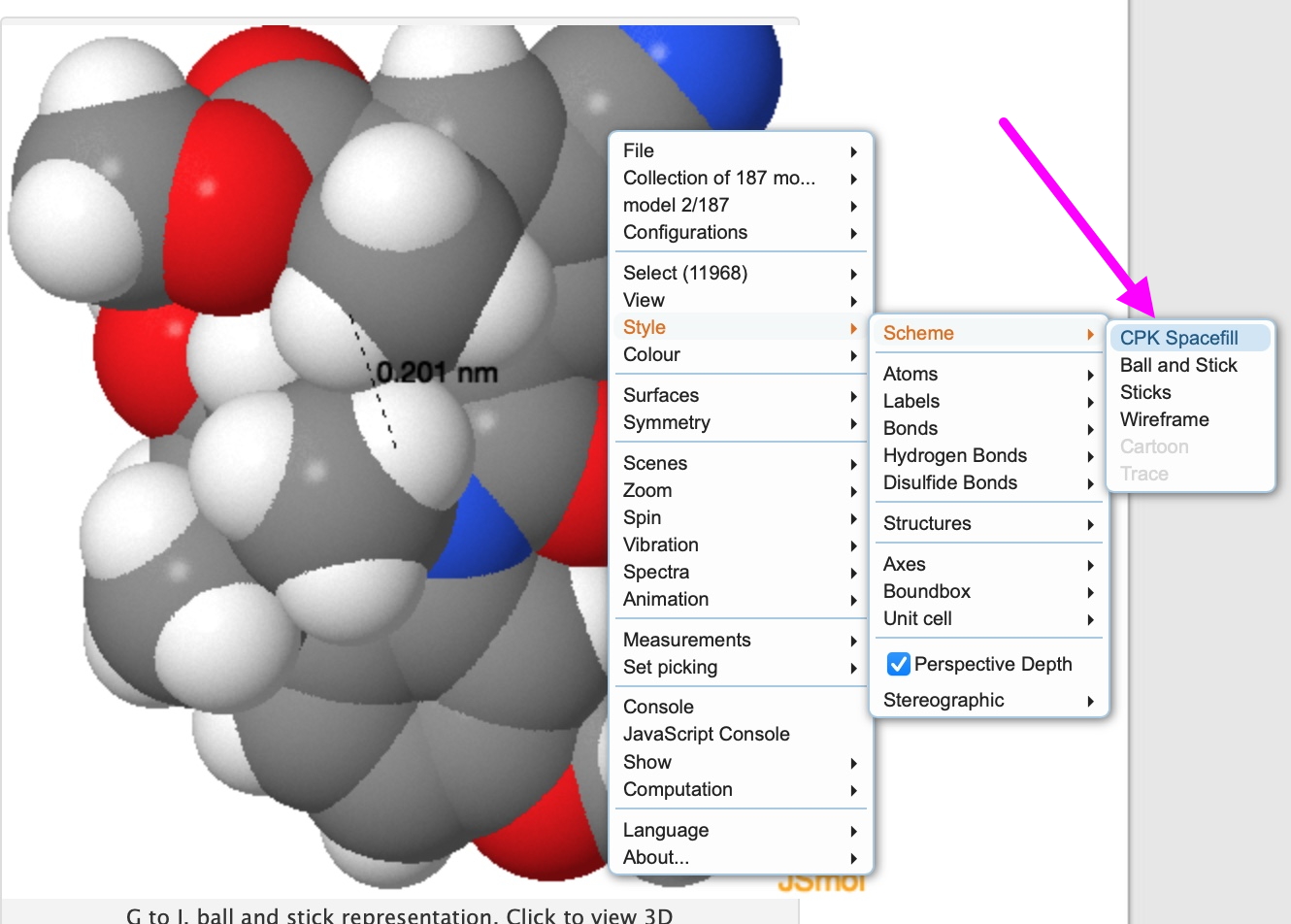
‡My thanks to Jeff Seeman and Dean Tantillo for contacting me about this, inspiring the above revisitation and much interesting discussion.[3]. †As noted elsewhere on this blog, H…H contacts as short as 1.5Å have been measured experimentally. ♥To turn the 3D view of the molecule into a spacefill model, right-click in the model window and invoke Scheme/CPK Spacefill as shown below:
The post has DOI: 10.14469/hpc/7807
Author
References
- R.B. Woodward, and R. Hoffmann, "Stereochemistry of Electrocyclic Reactions", Journal of the American Chemical Society, vol. 87, pp. 395-397, 1965. https://doi.org/10.1021/ja01080a054
- R.B. Corey, and L. Pauling, "Molecular Models of Amino Acids, Peptides, and Proteins", Review of Scientific Instruments, vol. 24, pp. 621-627, 1953. https://doi.org/10.1063/1.1770803
- D.J. Tantillo, and J.I. Seeman, "On the Structural Assignments Underlying R. B. Woodward's Most Personal Data That Led to the Woodward–Hoffmann Rules: Subramania Ranganathan's Key Role and Related Research by E. J. Corey and A. G. Hortmann", Chemistry – A European Journal, vol. 27, pp. 7000-7016, 2021. https://doi.org/10.1002/chem.202004790