Five years back, I speculated about the mechanism of the epoxidation of ethene by a peracid, concluding that kinetic isotope effects provided interesting evidence that this mechanism is highly asynchronous and involves a so-called “hidden intermediate”. Here I revisit this reaction in which a small change is applied to the atoms involved.
Below are two representations of the mechanism. The synchronous mechanism involves five “curly arrows”, two of which are involved in forming a bond between oxygen and carbon, and three of which transfer a proton to the group X (X=O). The second variation asynchronously stops at the half way stage to form a pseudo ion-pair (the “hidden intermediate”) and the proton transfer only occurs in the second stage. If the ethene is substituted with deuterium, experimentally an inverse kinetic isotope effect is observed, which provides strong evidence that at the transition state, no proton transfer is occurring

Before I go on, I should say that you will not find the mechanism as shown in either variation above in very many text books, which tend to practice “curly arrow economy” by employing only four arrows. I will not pursue this aspect here, except to note that as drawn above, the synchronous mechanism resembles that of a pericyclic reaction in a variation known as coarctate, as I noted in the original post (DOI: 10.14469/hpc/4807).
Now I introduce a veritable variation into this reaction, known as Payne epoxidation[1],† which replaces the peracid with a reagent generated by adding hydrogen peroxide to a nitrile to generate a transient species which can be represented by X=NH above. How does this change things? The model below also uses propene rather than ethene (M062X/Def2-TZVPPD/SCRF=dichloromethane).‡ This transition state (ΔG298 31.3 kcal/mol) shows two C-O bond formations, and as before the proton is clearly not yet transferred to the nitrogen (X=NH). Because of this asynchrony, the reaction could also be called a coarctate pseudo-pericyclic reaction.
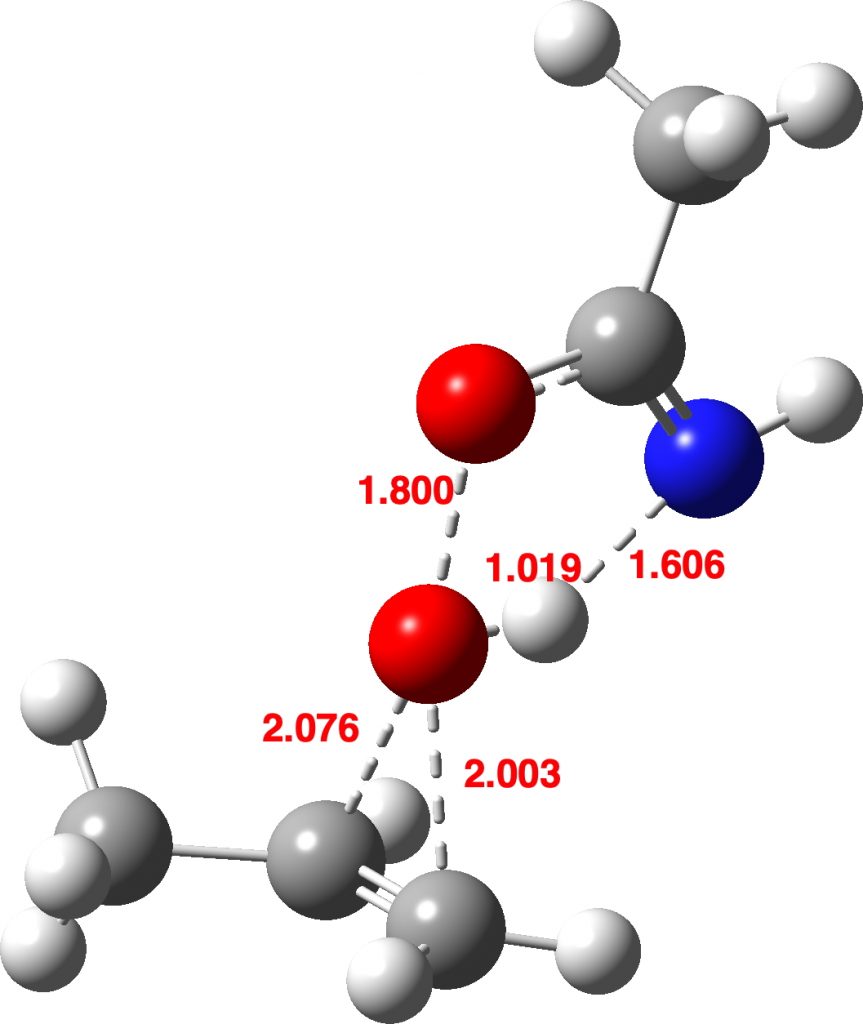
Asynchronous concerted mechanism. Click for 3D
However, the proton transfer is nonetheless part of a concerted mechanism, as shown by the IRC profile. 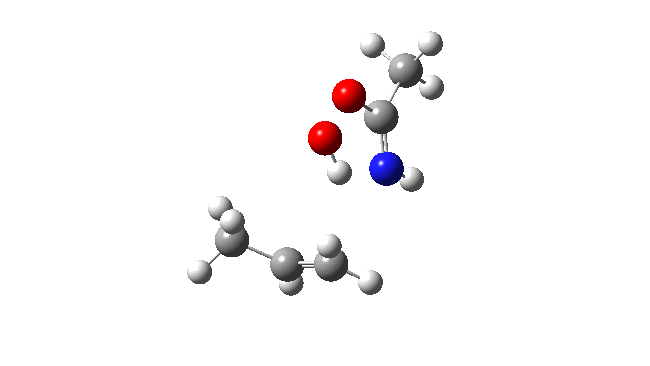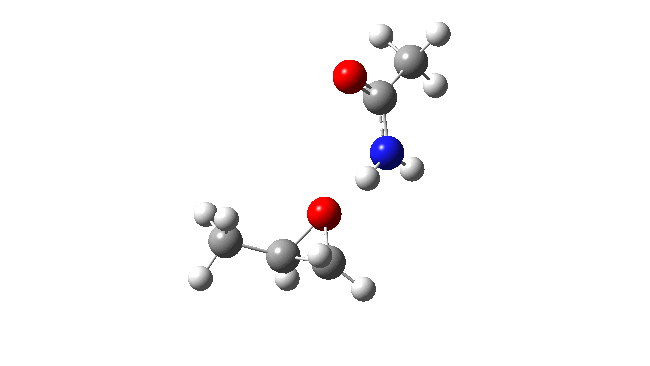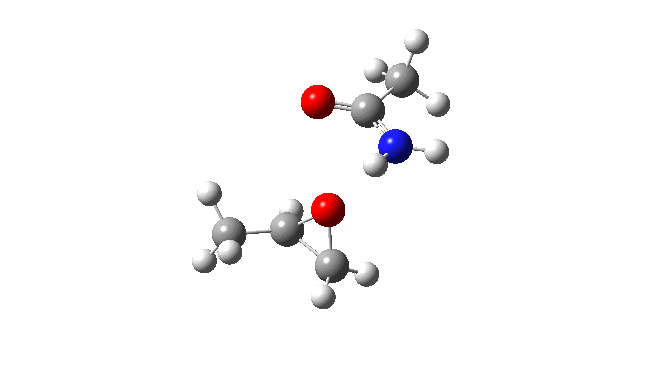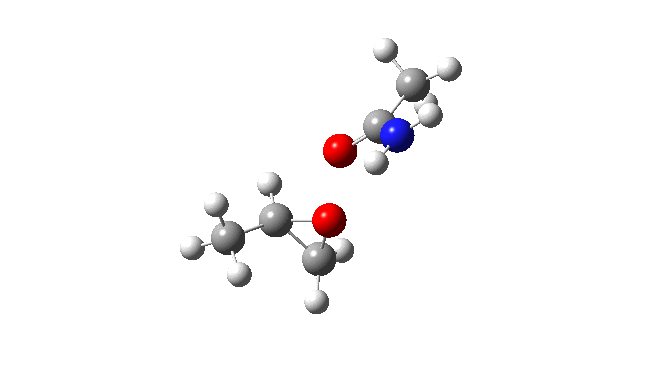

The gradient norm most clearly shows the “hidden ion-pair intermediate” at IRC = -1, and the proton transfer only occurs after this point is passed.
This is even more spectacularly illustrated with a plot of dipole moment along the IRC; 
In truth, no real differences are yet revealed between the Payne reagent and the peracid. In fact, this is a real surprise, since the NH of the Payne reagent should be very much more basic than the carbonyl oxygen of the peracid. But more exploration of the potential energy surface reveals another transition state!
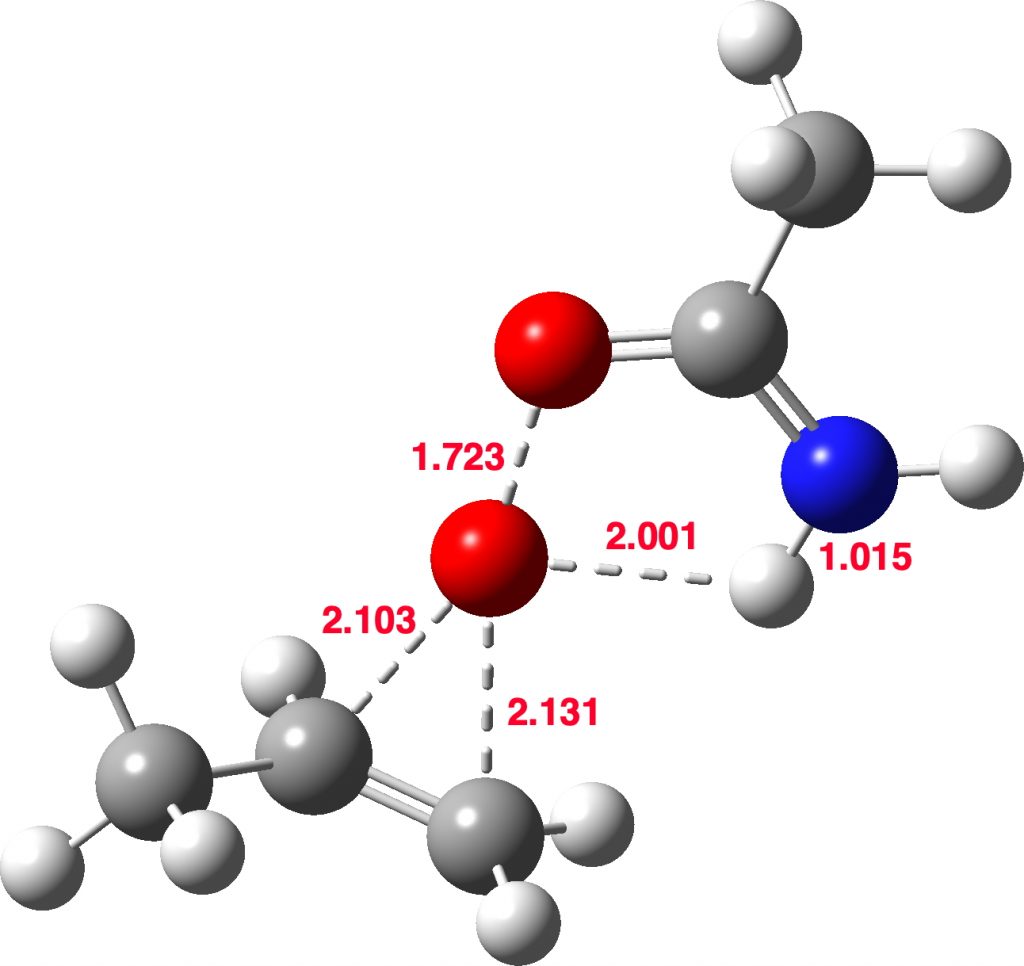
Stepwise mechanism. Click for 3D
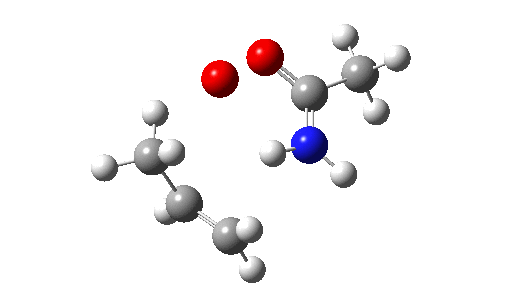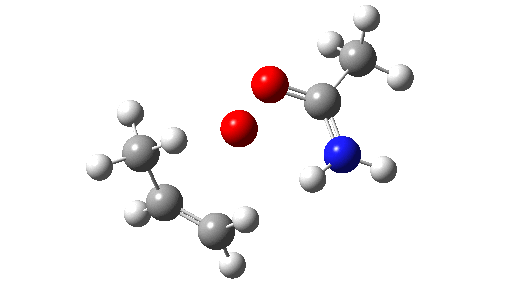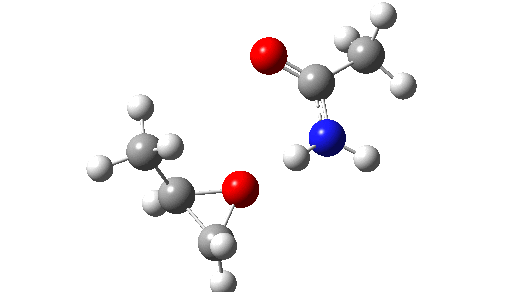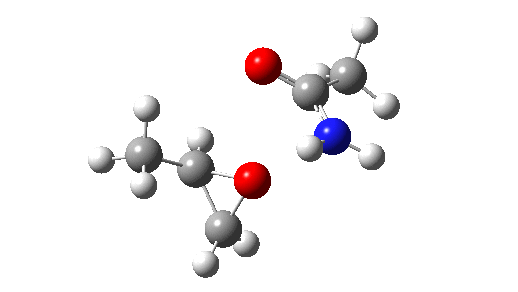 This is seen forming the two C-O bonds AFTER the proton transfer from oxygen to nitrogen. It is 4.2 kcal/mol lower than the first transition state, which corresponds to the scheme below.
This is seen forming the two C-O bonds AFTER the proton transfer from oxygen to nitrogen. It is 4.2 kcal/mol lower than the first transition state, which corresponds to the scheme below.

The new ion-pair shown above is 7.1 kcal/mol higher than the previous reactant, but is so much more basic than before that the overall activation energy is indeed lowered. Two distinctly separate IRCs can be constructed for this alternative, the first a pure proton transfer (not shown) and the second a pure C-O bond forming process (below). This second step is both concerted and almost purely synchronous.

So now we see how a small change to the reactant molecules (X=O to X=NH) can induce a reaction for which two quite different mechanisms can operate, an asynchronous one albeit with a hidden intermediate and a fully stepwise one in which a quite different, but this time real, intermediate is involved. Nevertheless for both the peracid mechanism and the peroxyimine variation shown here, the proton transfer is NOT involved in the rate limiting step. So for this variation too, inverse kinetic isotope effects would be expected.
‡FAIR data for the calculations at DOI: 10.14469/hpc/4909 †Thanks Ed for pointing this out.
Author
References
- G.B. PAYNE, P.H. DEMING, and P.H. WILLIAMS, "Reactions of Hydrogen Peroxide. VII. Alkali-Catalyzed Epoxidation and Oxidation Using a Nitrile as Co-reactant", The Journal of Organic Chemistry, vol. 26, pp. 659-663, 1961. https://doi.org/10.1021/jo01062a004
Tags: Chemical kinetics, chemical reaction, Chemistry, Deuterium, Isotope effect, Kinetic isotope effect, Natural sciences, Organic chemistry, overall activation energy, pericyclic reaction, Physical organic chemistry, Physical sciences, potential energy surface, Rearrangement reactions
IMHO, I think the term “hidden intermediate” is somewhat misleading. In contrast to a genuine intermediare as a local minimum on the PES, the “hidden intermediate” is not a minimum, and only characterized by a local minumum on the curve of force (or other quantities other than the potential energy).
Another question: how could you relate the appearance of a local minimim of the force along the IRC to the synchronous mechanism? Is there any chemcial meaning for this relationship? I’m not familiar with this tipic. Sorry for asking such a trivial question.
A concerted mechanism suggests there are no true minima along a reaction path other than the defined reactant and product. A synchronous concerted mechanism tends to imply that there are no “hidden” intermediates either, although true synchrony of all the forming and cleaving bonds can rarely be achieved unless imposed by symmetry. A stepwise reaction accepts that there will be one ore more true minima along a path connecting the defined reactant and product.
The term “hidden” was introduced by Dieter Cremer and is now accepted. It indeed carries the meaning that it is is only a local minimum in the curve of the force, but also carries chemical significance in as much as inspection of the geometry at the local minimum in the curve of the force can help one decide how to stabilize it out so that it might become a true (as opposed to hidden) minimum. Or indeed destablize it out so that loses this feature.
Thank you for the explanation. Did you suggest that one can stabilize the hidden intermediate out by varying the solvent or introducing some functional groups so that the concerted mechanism becomes truly stepwise? If I understood correctly, I would suggest to term the so-called hidden intermediate “secondary intermediate” for it is characterized by the second derivative of the potential energy.
In any case, I will go read Dieter Cemer’s article carefully.