Chloroform, often in the deuterated form CDCl3, is a very common solvent for NMR and other types of spectroscopy. Quantum mechanics is increasingly used to calculate such spectra to aid assignment and the solvent is here normally simulated as a continuum rather than by explicit inclusion of one or more chloroform molecules. But what are the features of the hydrogen bonds that form from chloroform to other acceptors? Here I do a quick search for the common characteristics of such interactions.
- This first search (R < 0.05, no errors, no disorder) is for interactions from the CH… O, and is a plot of that distance against the angle subtended at the oxygen.
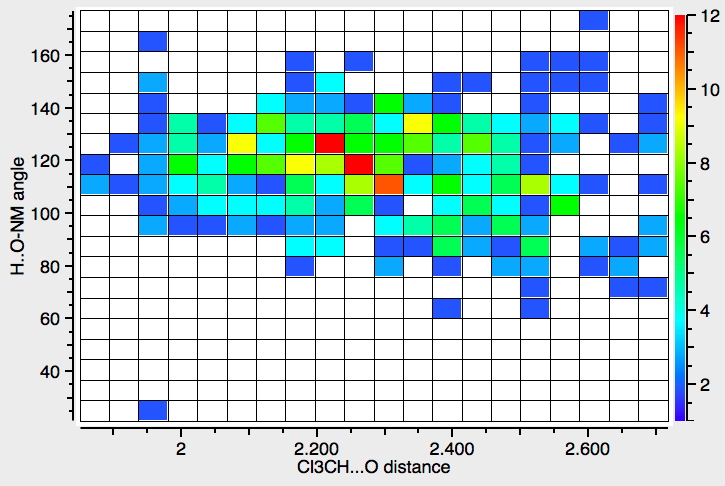
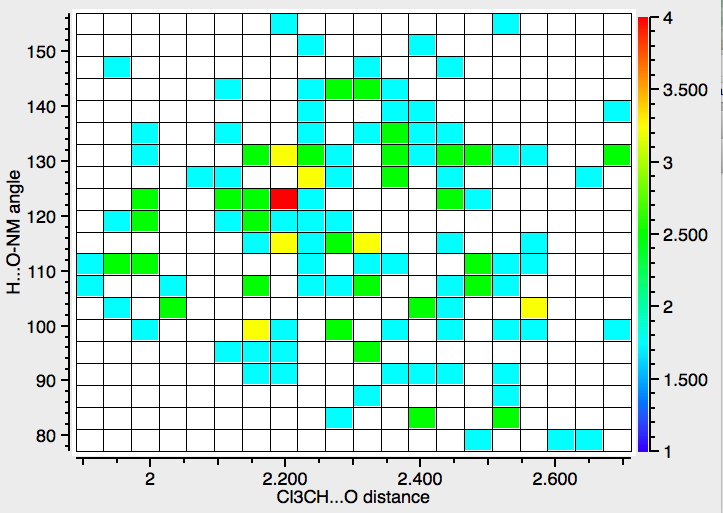
Note that there are not that many crystalline examples. The “hotspot” is at a distance of ~2.3Å, but real examples down to 1.9Å exist. The angle subtended at the oxygen is close to 120° (the angle subtended at the hydrogen is always close to 180°). The plot below constrains the search to data collected below 140K to reduce the thermal noise in the measurements, with the hotspot shortening slightly to 2.2Å.
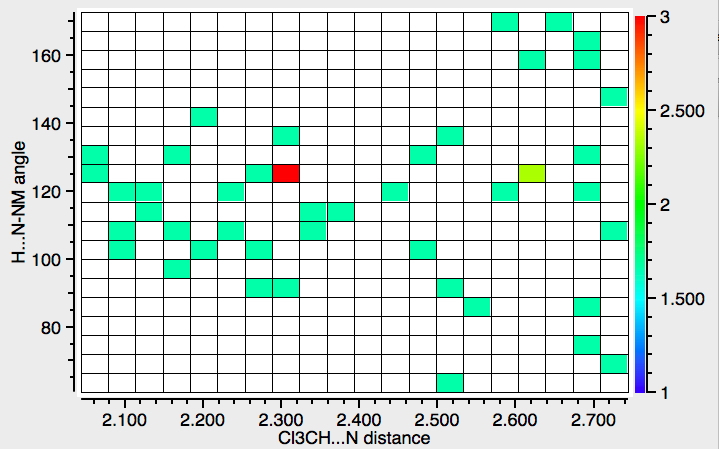
- The next search is for interactions to N rather than O (T < 140K). There are rather fewer hits, but again with similar features.
- Finally, an attempt to see if there is a correlation between the C-H length and the H…O length.
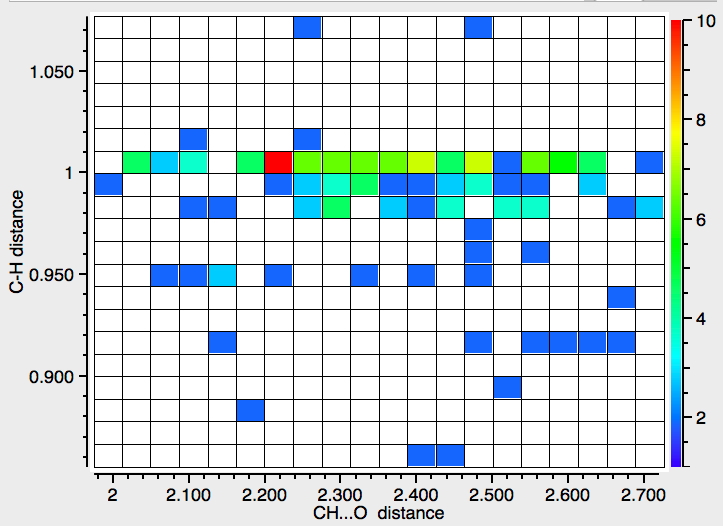
This has odd characteristics, which suggests that in most cases the C-H distance is not measured from the diffraction data but simply “idealised” (and which therefore renders this plot meaningless). Unless its been added recently, it is not possible to specify in the search how the hydrogen positions have been refined, if at all and hence to restrict the search only to those structures where the C-H distance is meaningful.
In the last ten years or so, great progress has been made in assigning experimental spectra with the help of quantum calculations. This is true of chemical shifts in NMR, but especially so for chiroptical measurements such as ORP, ECD and VCD. Given that explicit hydrogen bonds can introduce anisotropy into the otherwise isotropic solvent continuum, it might be worth including perhaps one chloroform molecule into these calculations, especially if the CH…O distance is <2Å (which suggests it is fairly strong). If nothing else, chloroform is rather big and might exert effects based on dispersion attractions or steric repulsions as well as the H-bonding.
Author
Tags: chemical shifts, Chloroform, Deuterated chloroform, Deuterated methanol, Hydrogen bond, Nuclear magnetic resonance, spectroscopy
So, your conclusion is that chloroform can form H-bonding almost as good as other typical H-bonding molecules. If this is true, have you tried with CHF3, where the H-bonding effect should be more, and there will be less steric effect.
A good suggestion! A quick search of CCDC (using the updated database with >1 million structures!) reveals no entries involving close contacts to the H of CHF3. I rather suspect this is because CHF3 is a gas and does not survive in solid structures. There is just one entry with CHF3 (DOI: 10.5517/ccdc.csd.cc1zwnh0) but with no apparent H-bonding occurring.
There are four examples of hydrogen bonding to CHBr3, with e.g. H…O distances of ~2.1Å. There are none for CHI3.
Looking recently for structural criteria for H-bonds in general has been driving me nuts. You touch above on a big problem: H-atom positions are generally designated arbitrarily. As a consequence, it seems to me that for a Z–H•••X interaction, the Z-H and X•••H distances are unreliable indicators. Many papers at least tell you what the Z–X distance is, which is probably a fair guide. Your opinion ?
Tony,
Idealized H-atom positions are largely a historical issue. Nowadays the H-positions positions are normally refined, along with all other atoms. Moreover, if an atom position is idealized, it is normally fairly close to the eventual refined position. The Z-X distance is of course more reliable. Ideally, if a H-position is refined, it should also be done at low temperatures, to reduce uncertainty of thermal motions. Finally, when searches of such systems are carried out in the CSD database, it is relatively easy to “correct” the position of a refined H atom by a fairly reliable amount, so that the overall trends in H-position are more accurate.