In the preceding post, I discussed the reaction between mCPBA (meta-chloroperbenzoic acid) and cyclohexanone, resulting in Baeyer-Villiger oxidation via a tetrahedral intermediate (TI). Dan Singleton, in whose group the original KIE (kinetic isotope measurements) were made, has kindly pointed out on this blog that his was a mixed-phase reaction, and that mechanistic comparison with homogenous solutions may not be justified. An intriguing aspect of the (solution) mechanism would be whether the TI forms quickly and/or reversibly and what the position of any equilibrium between it and the starting ketone is. This reminded me of work we did some years ago,[1] and here I discuss that.
It involved the addition of phenyl hydroxylamine, PhNHOH to acetyl cyanide at 215K. Because the CN group is poor at leaving, the tetrahedral intermediates do not collapse and instead accumulate in seconds to the point of becoming detectable by NMR (both N-C and O-C isomers). The position of the equilibrium clearly favours the TI rather than the starting materials. In another context, both the rate of reaction and the equilibrium can be driven towards the TI by the application of pressure.[2] Hydroxylamines are known to be super nucleophiles, enhanced by the so-called α-effect from buttressing of adjacent lone pairs on the N and O. This reminds that a peracid also should exhibit a related α-effect; it should be a better nucleophile than a normal carboxylic acid. So I decided to take the TI formed from cyclohexanone and mcPBA and look at the NBO orbitals, which should tell us about the anomeric effects present in this TI, and in particular if they might be larger than normal (which could be equated with greater stability for the TI). Here are the relevant NBO energies.[3]
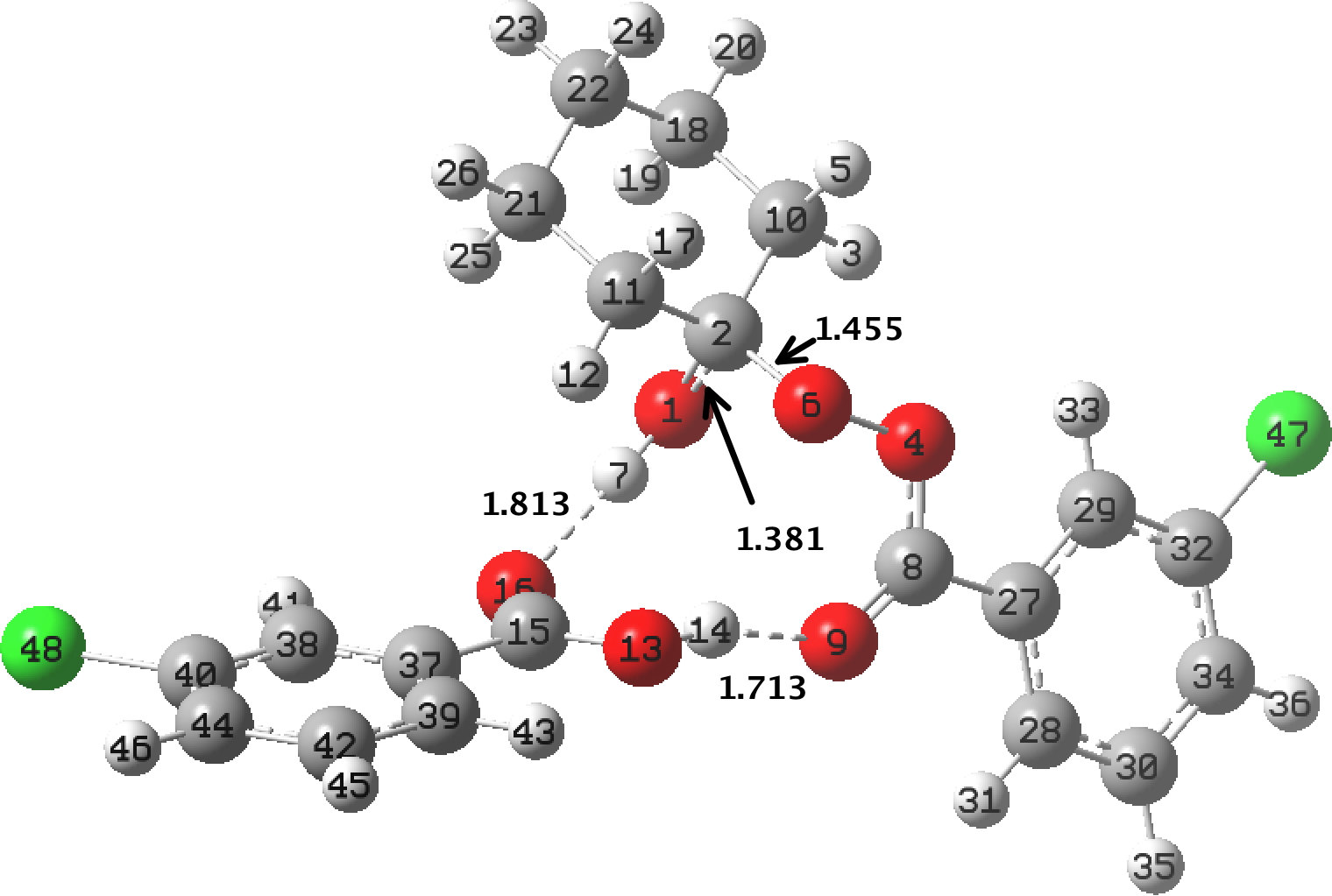
- The conventional anomeric effect in O-C-O manifests as a E(2) perturbation energy of ~16-18 kcal/mol between one oxygen lone pair and the antibonding C-O orbital. There are two combinations, and these are normally similar in energy.
- For the system above, the O1-C2-O6 interaction is 25.6 kcal/mol, much larger than normal, but partially counterbalanced by:
- O6-C1-O2 =13.0 kcal/mol which is a little lower than normal. This is overall an unusually strong anomeric effect for the O-C-O motif!
- The energetic asymmetry is matched by the two computed bond lengths, 1.381Å for the larger interaction and 1.455Å for the smaller. The pseudo-α-effect has desymmetrized the anomeric effect, but nevertheless strengthened it overall.
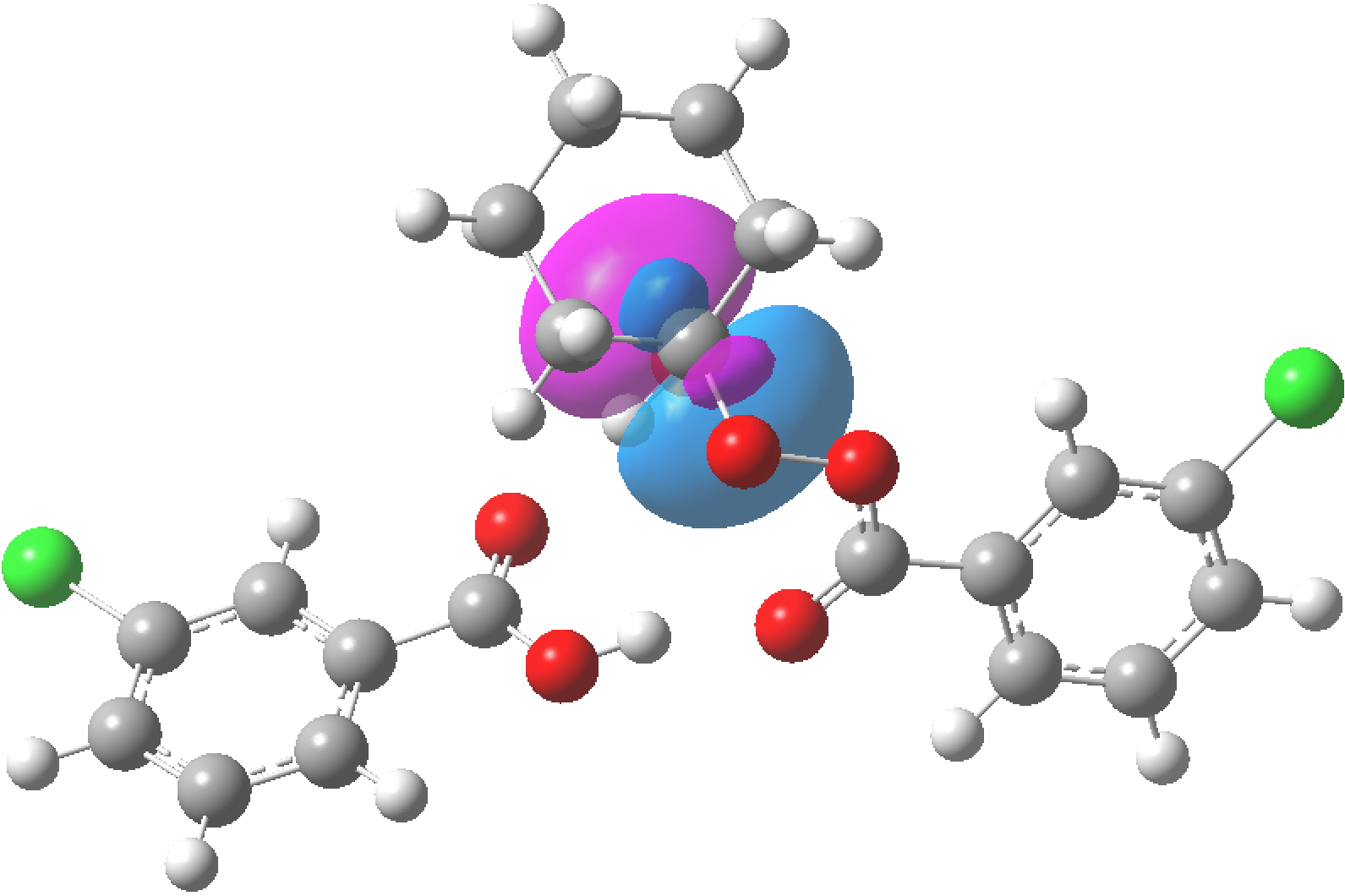
NBO 103 for O1(Lp)
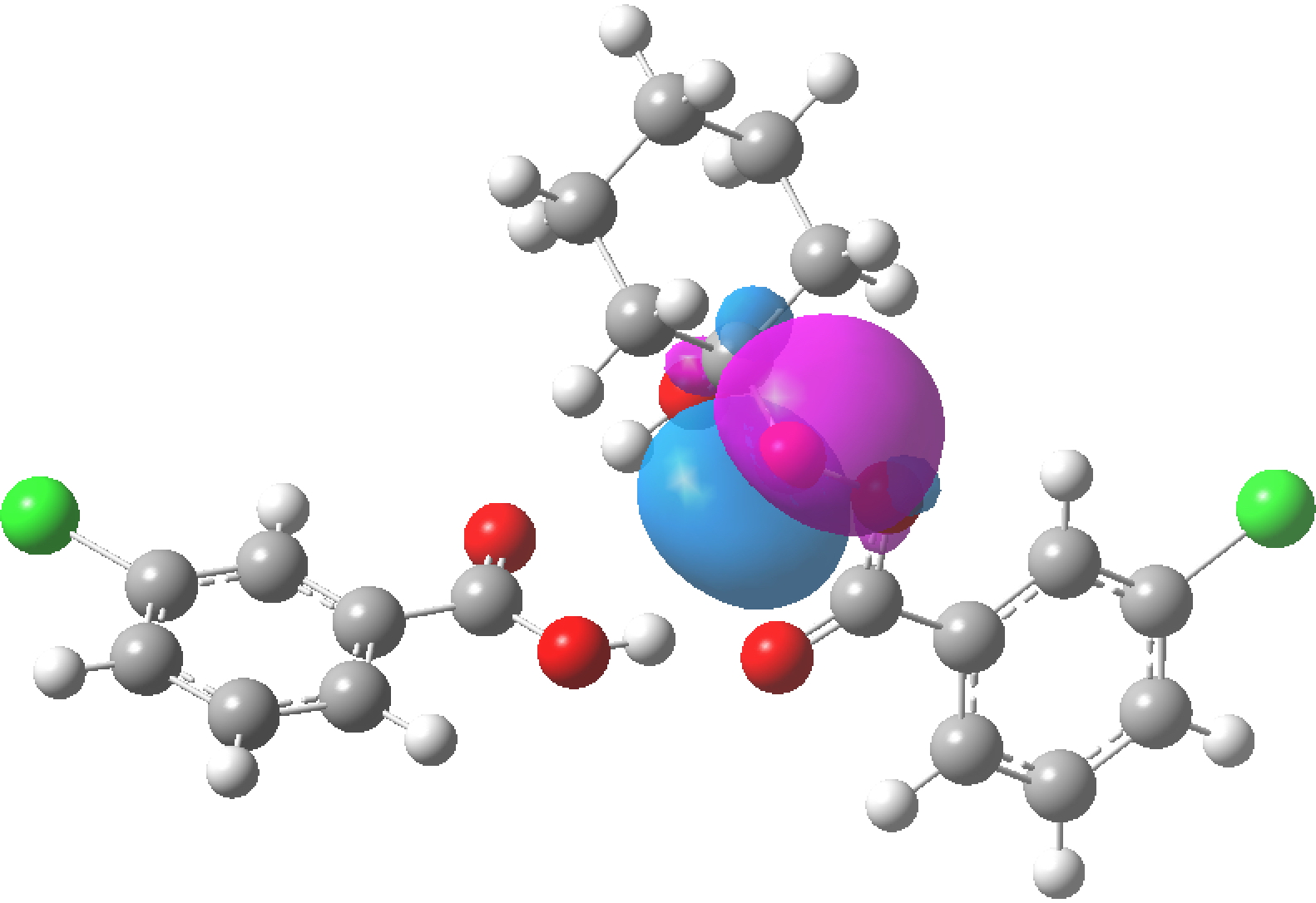
NBO 97 for O6(Lp)
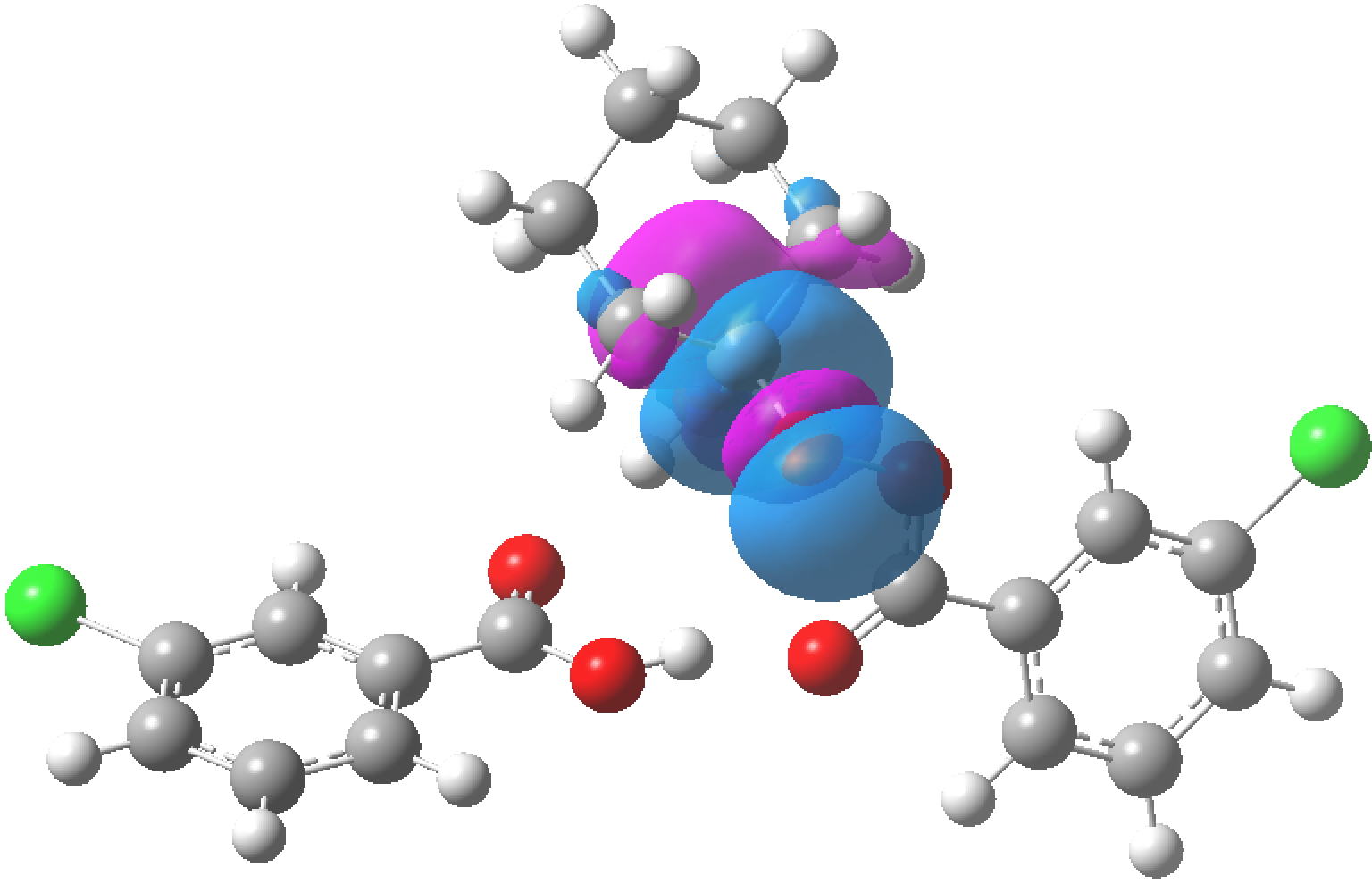
NBO 123 for C2-O6 antibonding σ*orbital
One concludes that the asymmetric anomeric effect makes the TI resemble the reactants. The transition state leading to the TI must be even earlier. In this context, I note that the (mixed phase) 13C effect reported for the carbonyl by Singleton and Szymanski[4] was quite a large one for carbon (1.045-1.051), a magnitude which argues against a very early transition state under these conditions. But the calculated value for a homogenous solution state model of ~1.023 is certainly more in accord with an early transition state.
Finally, a search of the CSD reveals 12 molecules containing either a O-O-C-O-O or a O-C-O-O sub unit This one[5] shows a bis HO-C-O-O-C-OH structure at room temperature; these species need not be unstable! There are none however with Ac-O-O-C-O. And of course the potent antimalarial artemisinin contains a O-O-C-O-C-O-Ac unit, for which stereoelectronic effects may also be important.
Author
References
- A.M. Lobo, M.M. Marques, S. Prabhakar, and H.S. Rzepa, "Tetrahedral intermediates formed by nitrogen and oxygen attack of aromatic hydroxylamines on acetyl cyanide", The Journal of Organic Chemistry, vol. 52, pp. 2925-2927, 1987. https://doi.org/10.1021/jo00389a050
- N.S. Isaacs, H.S. Rzepa, R.N. Sheppard, A.M. Lobo, S. Prabhakar, and A.E. Merbach, "Volumes of reaction for the formation of some analogues of tetrahedral intermediates", Journal of the Chemical Society, Perkin Transactions 2, pp. 1477, 1987. https://doi.org/10.1039/p29870001477
- H.S. Rzepa, "C 20 H 20 Cl 2 O 6", 2015. https://doi.org/10.14469/ch/191327
- D.A. Singleton, and M.J. Szymanski, "Simultaneous Determination of Intermolecular and Intramolecular <sup>13</sup>C and <sup>2</sup>H Kinetic Isotope Effects at Natural Abundance", Journal of the American Chemical Society, vol. 121, pp. 9455-9456, 1999. https://doi.org/10.1021/ja992016z
- A. Kobayashi, Y. Ikeda, K. Kubota, and Y. Ohashi, "Syntheses and crystalline structures of several aldehyde peroxides as new flavor compounds", Journal of Agricultural and Food Chemistry, vol. 41, pp. 1297-1299, 1993. https://doi.org/10.1021/jf00032a025
Tags: Anomer, Anomeric effect, Carbohydrate chemistry, Carbohydrates, Chemistry, Dan Singleton, homogenous solutions, Ketone, Meta-Chloroperoxybenzoic acid, Organic chemistry, Tetrahedral carbonyl addition compound
Hello Henry,
You mention the alpha effect as the origin of the enhanced nucleophilicity of hydroxylamine. I remember a paper from a long time ago (1983?) which explored nucleophilic addition in the gas phase. These authors found that the effect disappeared, leading them to question its reality. Do you have any update on the current sate of play?
The alpha effect appears not to be a well-characterised search term. Thus Scifinder reports 1983 “hits” on the phrase “alpha effect in nucleophilic additions”. Perhaps there is gold there, but I am not going to scan all these hits to find it. Wikipedia states “The effect is well established with many theories to explain the effect but without a clear winner.” By this they probably mean in solution.
Scifinder does find this article from 2008 doi:10.1039/b803734e entitled “Reactions of the hydroperoxide anion with dimethyl methylphosphonate in an ion trap mass spectrometer: evidence for a gas phase α-effect” which perhaps can be taken as state of the play (in 2008). But the effect was relatively small, amounting to ~2 kcal/mol. Very probably any effect induced by R-O-O is going to be smaller than eg the corresponding R-NH-O motif. Still, for this specific system it does appear to be real in the gas phase.
However, two other more recent articles; doi:10.1021/jp407698a and 10.1021/ja9012084 suggest that indeed the effect is induced by solvent. The first studies microsolvation (with one methanol molecule), finding just this one solvent molecule can induce the effect. This is an interesting comment “In addition, solvent interactions for HOO(-) are quite different from those with the normal nucleophiles at the transition state, indicating that differential solvation may well contribute to the α-effect.”
From this we might conclude that when constructing transition state models, perhaps they should be routinely micro solvated. Certainly in the Baeyer-Villiger reaction, microsolvation played a crucial role in the proton transfers.