In London, one has the pleasures of attending occasional one day meetings at the Burlington House, home of the Royal Society of Chemistry. On November 5th this year, there was an excellent meeting on the topic of Challenges in Catalysis, and you can see the speakers and (some of) their slides here. One talk on the topic of Direct amide formation – the issues, the art, the industrial application by Dave Jackson caught my interest. He asked whether an amide could be formed directly from a carboxylic acid and an amine without the intervention of an explicit catalyst. The answer involved noting that the carboxylic acid was itself a catalyst in the process, and a full mechanistic exploration of this aspect can be found in an article published in collaboration with Andy Whiting's group at Durham.[1] My after-thoughts in the pub centered around the recollection that I had written some blog posts about the reaction between hydroxylamine and propanone. Might there be any similarity between the two mechanisms?

That mechanism can be represented as above, which (as per the hydroxylamine mechanism) comprises three transition states and two intermediates. The original study[1] reported just the one TS1. Editing out the starting coordinates from the PDF-based supporting information (the process is not always easy) enabled an IRC (intrinsic reaction coordinate) for TS1 to be easily computed.[2]
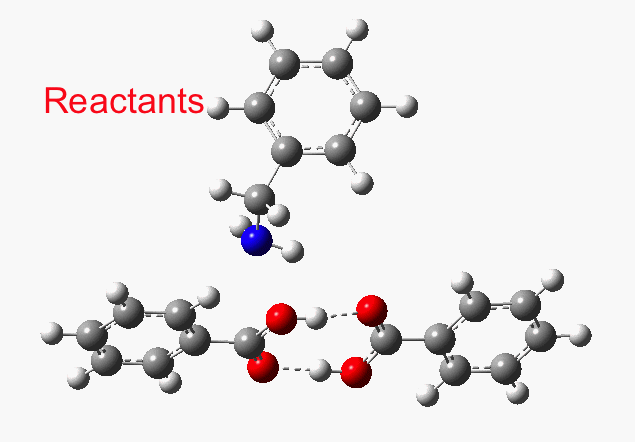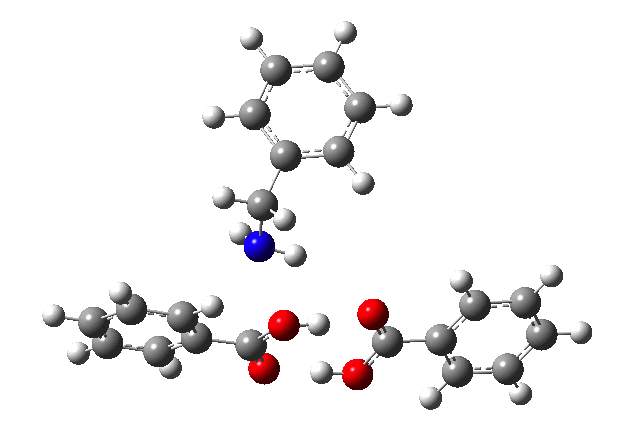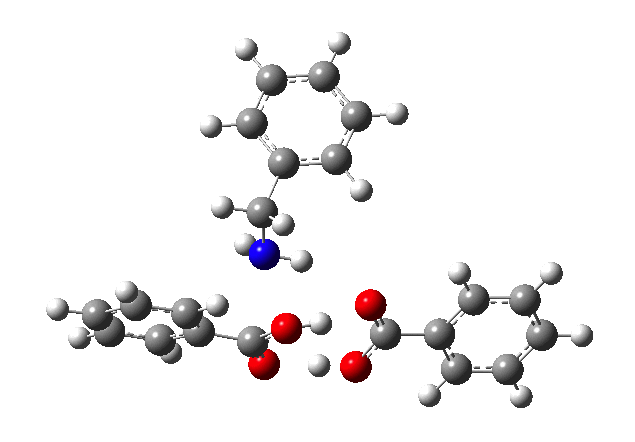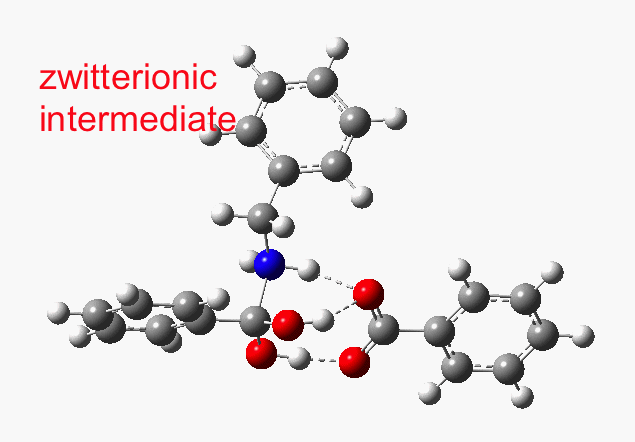

This reveals that TS1 is not the complete story, there is still much of the reaction left to complete. The energy profile is charted (using the ωB97XD/6-311G(d,p/SCRF=p-xylene method) according to the scheme above as reactants ⇒ TS1 ⇒ Intermediate 1 ⇒ TS2 ⇒ Tetrahedral intermediate ⇒ TS3 ⇒ products. Computed properties for this more detailed pathway are transcluded here from the digital repository[3] and appear at the end of this post.
- TS1 yields what might be called a zwitterionic intermediate. However, this has a relatively small dipole moment (5.7D). Thus, against accepted wisdom, such apparently ionic intermediates CAN be involved in reactions occurring in non-polar solvents!
- TS2 is rather unexpected, involving synchronous proton transfer coupled to anomerically related C-OH bond rotation. This rotation changes the anomeric interactions with the adjacent substituents; in my experience I have never before seen a reaction mode quite like this one!
- TS3 collapses the tetrahedral intermediate by synchronous proton transfer and C-O bond cleavage, and is (in this model) the rate determining step. The free energy barrier corresponds to a half-life at 298K of about half an hour.
- The product is calculated as exoenergic with respect to reactants,; the reaction does drive to form an amide (and any catalysis of course will not influence that final outcome, only its kinetics).
If you read the original article[1] you will realise the above only scratches the surface of the many fascinating properties of this apparently very simple reaction. Thus, not addressed above is why amides are only formed in certain solvents (xylene for example) but not others. The solvent may have a specific role to play which is not modelled simply by its continuum dielectric or its boiling point. There is much else that could be said.
Author
References
- H. Charville, D.A. Jackson, G. Hodges, A. Whiting, and M.R. Wilson, "The Uncatalyzed Direct Amide Formation Reaction – Mechanism Studies and the Key Role of Carboxylic Acid H‐Bonding", European Journal of Organic Chemistry, vol. 2011, pp. 5981-5990, 2011. https://doi.org/10.1002/ejoc.201100714
- H.S. Rzepa, "C21H21NO4", 2014. https://doi.org/10.14469/ch/74636
- H.S. Rzepa, "A computed mechanistic pathway for the formation of an amide from an acid and an amine in non-polar solution.", 2014. https://doi.org/10.6084/m9.figshare.1235300
Tags: Andy Whiting, Dave Jackson, dielectric, Durham, energy profile, free energy barrier, London, non-polar solution, PDF, Royal Society of Chemistry
How does this avoid making the amine carboxylate and getting stuck there?
@Mark: It is all reversible, so no problem in the long run. The acid-base-equilibrium just reduces the available free acid/base concentrations and makes kinetics slow.
The following experiment has been performed by Menschutkin in the 1880’s (http://onlinelibrary.wiley.com/doi/10.1002/prac.18820260113/abstract):
Aniline and acetic acid are mixed in 1:1 molecular ratio and amide formation was followed at different temperatures:
1. At 155 °C: 1 h = 58%, 2 h = 66%, 4 h = 74%, 8 h = 77%, 24 h = 79%, 120 h = 80%
2. At room temperature: 2 d = 1.1%, 5 d = 5.4%, 12 d = 8.2%, 31 d = 12%, 59d = 26%, 137 d = 44%.
I have performed a similar experiment between aniline and mercaptoacetic acid (neat) and found:
8 d = 24%, 19 d = 32%, 45 d = 49%, 155 d = 72%.
The expeirments suffer from the unclear role of water (did it leave the system in Menschutkins setup? Probbably not, for the conversion stopped at 80% in his case).
It is a challenge for catalysis research to find a catalyst that works at r.t. (without addition of molecular sieves)!
Re: Mark Penick. Your question is central. In fact in polar solvents the carboxylic acid and amine do indeed simply form an ammonium carboxylate salt, thus inhibiting the reaction. However in xylene, which is non-polar, this ionic system is not stable. I did try to compute its energy, but at what I thought were reasonable geometries for the close ion pair, the proton always transferred back to form neutral components. In xylene, there is no opportunity to form a solvent-separated ion pair (at least that is what the conventional wisdom would have). I have not tried adding one or more xylenes to the model, but that would be worth doing.
Furthermore, xylene-water forms a eutectic, and this serves to boil off the water at relatively low temperatures, thus shifting the equilibrium towards amide. This combination of non-polarity and favourable eutectic characteristics is what makes the xylene system relatively unique.
You can get lots more information from Dave Jackson’s slides, where he covered these points extensively.
re Lukas: Yes, removal of water does indeed determine whether the reaction goes to completion, and yes Dave Jackson in his talk did discuss the role of molecular sieves in this process. On an industrial scale, these apparently are a relatively expensive way of removing water. And yes, the message that came from Dave’s slides is that this is indeed the challenge, to find a catalyst that works at r.t. and is “green”.
Regarding the calculations, they did predict a slightly exoenergic outcome for the reaction. In part this is because of the formation of a stable cyclic hydrogen bonded product, very similar in fact to the acetic acid dimer that is the starting point. And this is only possible with a primary amine to form that H-bond.
And (this is a real-time conclusion), as I wrote the previous paragraph, I thought I should check if this assertion was well founded in reality. A search of the Cambridge crystal database literally took 4 minutes, and here are two results. The first is a heat plot showing the distribution of such structures. The “hot spot” at distances of 1.8 and 2.1Å corresponds to the HO…O=C and NH…O=C H-bonds respectively. There appear to be about 100 examples of this motif.
Note that from this plot the scatter extends down to distances of 1.4 and 1.8Å. One would need to determine if these are real and not artefacts. If they are real, can we learn from them?
The second graphic below shows an explicit example of a cyclic trimer with this H-bond pattern.
Postscript. I have added a third graphic below, which is a re-run of the first with one change. In the search, the hydrogen positions are now normalised. This takes into account that X-ray scattering produces eg O-H distances which are about 0.1Å too short. This normalisation corrects this, with the result that the non-bonded hydrogen bonds get shorter.
This mechanism is actually quite familiar to me. It is basically the same that we found when we did QM/MM calculations on a bacterial peptide to explain spontaneous isopeptide bond formation where the carboxyl group of a GLU residue acts as a catalyst. It seems Nature likes to make amide bonds like this as well:
http://onlinelibrary.wiley.com/doi/10.1002/anie.201004340/abstract
Cheers, Ragnar
I actually recognize this mechanism. It is very similar to the mechanism that we figured out for isopeptide bond formation in a bacterial peptide a few years ago using QM/MM calculations:
http://onlinelibrary.wiley.com/doi/10.1002/anie.201004340/abstract
It seems Nature likes this way of creating amide bonds as well.