A staple of introductory undergraduate teaching in organic chemistry is Markovnikov’s rule, which states: “the addition of a protic acid HX to an alkene results in the acid hydrogen (H) becoming attached to the carbon with fewer alkyl substituents and the halide (X) group to the carbon with more alkyl substituents“. Shortly thereafter, students are exposed to the “anti-Markovnikov” addition of borane to e.g. 2-methylpropene. In order to achieve a consistent explanation for both reactions, I normally show students the following mechanism. Here I introduce a “reality check” to the first component of that mechanism (for the oxidative step, see this post).

The premise of this mechanism is that electronically at least, both the Markovnikov and the anti-Markovnikov additions actually arise from the same effect, which is to produce predominantly the more highly substituted intermediate carbocation. So the issue now is whether this intermediate, as might be invoked above for hydroboration, is real or whether it is simply an expediency to enable students to recognise the single electronic origin of both types of reaction (? above). Enter the ωB97XD/6-311G(d,p)/SCRF=thf procedure. Firstly, I note that the reaction is normally done with “stabilized” borane, via pre-coordination to thf.
Starting from either Markovnikov or anti-Markovnikov orientations, only one transition state could be found[1], meaning that the anti-Markovnikov preference is determined entirely by this transition state. The exit trajectories can presumably lead to either product, but favouring anti-Markovnikov. The transition state shown here was also used by Singleton[2] for a molecular dynamics study of the hydroboration of propene with BH3.thf. There, dynamics were invoked to explain why 10% of the product goes Markovnikov.
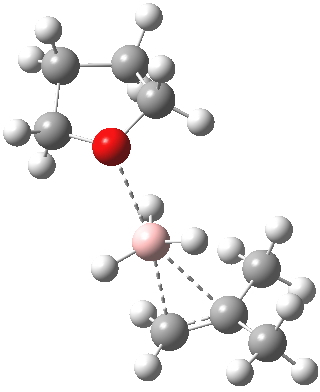
Transition state for addition of borane to butene. Click for 3D
The reaction with thf clearly occurs in two stages.[3] The first stage subsumes the transition state and is an SN2 like nucleophilic displacement at boron by the alkene, liberating thf as the leaving group. The second stage reveals an excellent example of a “hidden intermediate“, which could be described as the zwitterionic carbocation shown in the scheme above. Perhaps instead it is less classical than this, being closer to a non-classical bridged species involving an asymmetric π-complex between the borane and the alkene. At any rate, at IRC ~8, this hidden intermediate now decides to transfer a hydride to form the alkyl borane rather than to become an explicit intermediate.
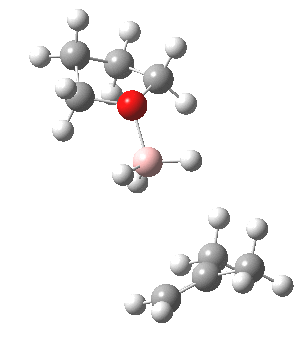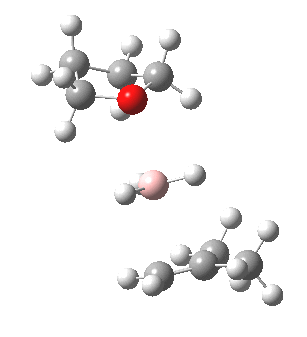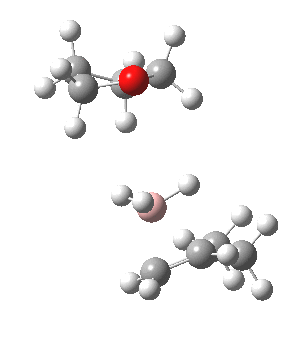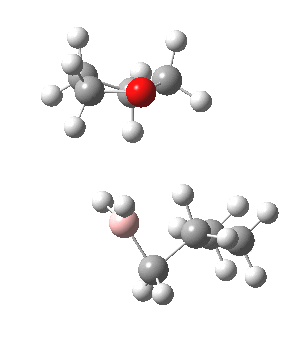



So ? turns out to be a hidden intermediate rather than an explicit one in this model. This hidden intermediate plays the role of the conventional transition state in its presumed determination of the regioselectivity of the hydroboration reaction. The IRC of course is a single trajectory; molecular dynamics will give a more statistical indication of the product distribution. Certainly, these “hidden intermediates” as key structures in mechanistic pathways are starting to turn up in more and more classical reactions.
Author
References
- H.S. Rzepa, "Gaussian Job Archive for C8H19BO", 2014. https://doi.org/10.6084/m9.figshare.949678
- Y. Oyola, and D.A. Singleton, "Dynamics and the Failure of Transition State Theory in Alkene Hydroboration", Journal of the American Chemical Society, vol. 131, pp. 3130-3131, 2009. https://doi.org/10.1021/ja807666d
- H.S. Rzepa, "Gaussian Job Archive for C8H19BO", 2014. https://doi.org/10.6084/m9.figshare.949689
Took me a while to figure out how but-2-ene could undergo any kind of regioselective reaction until I looked at the pictures and realized you meant isobutylene, aka 2-methylpropene.
Mea culpa. Corrected!
Do you really show the Lewis base coordinated to borane when you teach this mechanism to early years undergraduates? Can’t say I’ve ever seen it written down before but it is probably correct. We managed without http://www.chemtube3d.com/OrganoMainGroupChem(Boron)-Hydroboration(AddnBoronHydridesToAlkenes).HTML at a much reduced level of theory.
Nick: Well, without prior coordination, the “pre-complex” of borane + alkene lies in an extremely shallow minimum (it can be tricky to find it) and the barrier out of this minimum is very small. With pre-coordination to thf, the transition state changes very substantially in character, and now has a more reasonable barrier to reaction.
I agree that the text books do not show this, and it is also true most introductory level teaching comes straight from a text book!
Changing the topic slightly to MOOCs (massive open online courses). If indeed it is perfectly adequate to teach all introductory chemistry pretty much from “text-book” models, then one has to pose the question: why not all such teaching be done from the “best MOOC out there?” What could local delivery of notes transcribed in this manner add to the process?
My answer to that last question is that one should interpret even introductory chemistry using local expertise, and that way local teaching will have a very unique, differentiating flavour, perhaps in a manner that maps better with the other locally differentiated materials. I don’t think that a single, “definitive” way to teach any aspect of chemistry exists, and certainly there are no MOOCs the content of which have convinced me that it could even exist.
Of course, I realise that this might, to a text book author such as yourself, be a red flag to a bull!
Henry,
You may or may not be aware that Liverpool is in partnership with FutureLearn and, amusingly, I am one of the lead academics on the project. I have no plans to create a MOOC myself yet though.
I think it is useful to have a foundation for teaching provided by a textbook (ideally one that you have written to avoid unfortunate content!) and a website (ditto) but the idea that we could create a definitive monoculture that would render local input redundant is emphatically not part of my vision. Even my own colleagues depart from Clayden/ChemTube3D doctrine with regularity and I’m happy to support them.
The OERs/MOOCs that I am aware of have some good aspects but usually grate in other areas. In this internet age we have to learn wherever we can but I don’t see our day jobs becoming redundant any time soon.
In other parts of the world Organic Chemistry is a required course for those who are not destined to become chemists, never mind organic chemists, and for them, judging by the similarity between the available textbooks, a MOOC approach might cover the material. How effectively they would learn is another question.
In my 2nd semester organic chemistry lecture some time back, the coordinated THF was also shown in the transition state, though I cannot say I have kept remembering that aspect. We used printed lecture notes (as opposed to a textbook) that were updated each year and which clearly represented what Henry calls local expertise – I think they were optimized over several years by local professors and handed over to successors. Actually, I continue using part of this material in my own teaching now many years later, at a different university.
Concerning the “anti-Markovnikov” orientation (and Henry has already enclosed the term in quotation marks) I don’t like the term for hydroboration, since the original rule by Markovnikov was relating to HX additions, where X was “haloid” – which I interpret as an electronegative element – whereas boron is electropositive.
When I teach hydroboration I just state that the overall process (after oxidation) gives the product of formal anti-Markovnikov hydration of an alkene, as opposed to the acid-catalyzed hydration of alkenes, which is a typical example of Markovnikov regioselectivity.
In the local expertise of my undergraduate lectures, the historical version of Markovnikovs rule was contrasted with a “modern Markovnikov rule”, which said that the Markovnikov product is the one formed along the pathway via the more stable carbenium ion. In that sense, hydroboration could be regarded as displaying Markovnikov selectivity, as it is possible to draw a resonance structure of the transition state with a plus charge on the inner carbon and a minus on boron.
Whether such an application of the Markovnikov term is correct may certainly be debated – as in other reactions for which it was not designed for: Wacker oxidation, or alkyne hydration (which are often considered to display Markovnikov regioselectivity).