{kind=link}
{kind=link}
Catalysis with palladium metal or palladium complexes is established in organic synthesis. Perhaps the most important and widely used tasks that can be effected with Pd catalysts are C-H and C-O bond formation; but so far, no significant enantioselective asymmetric synthesis has emerged in these applications. C-C Bond forming cross coupling reactions, the Heck, Stille and Suzuki reactions, are of great importance for the connection of sp2 centers, e.g., diene synthesis. It was only recently shown by Shibasaki, Overman and others that the Heck reaction with cyclic alkenes and in Heck induced ring formations can be channeled towards chiral products with high enantioselectivity[1]. These are very recent, important achievements. The traditional battle field of Pd catalyzed asymmetric synthesis are C-C and C-N bond forming substitutions at allylic compounds. For a considerable time progress in this area was slow, but over the last few years dramatic improvements were achieved. Only the beginning of this development has been reviewed[2]. Here we give an account of our and related recent work of others in this area.
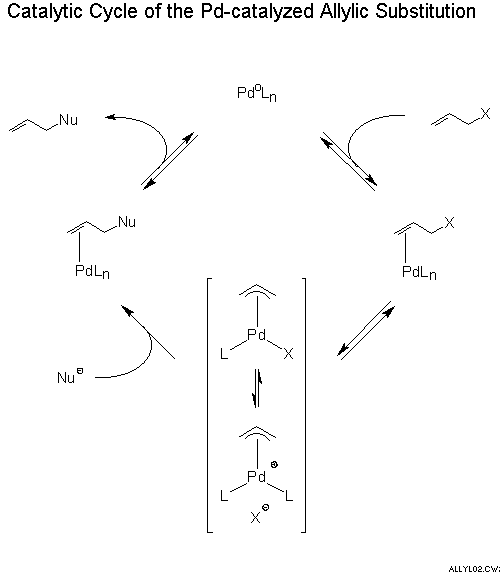
The catalytic cycle (Scheme 1) of a Pd catalyzed substitution involves first coordination of a Pd0 species to the double bond of an allylic system and then expulsion of the leaving group X to give a pi-allyl PdII intermediate which, depending on ligands L and counter ion X, can be a neutral or, presumed to be much more reactive, a cationic complex. A soft carbanion attacks at carbon. This reaction is usually irreversible and turnover determining. The resulting Pd0 olefin complex dissociates to yield the product and regenerate the catalyst.
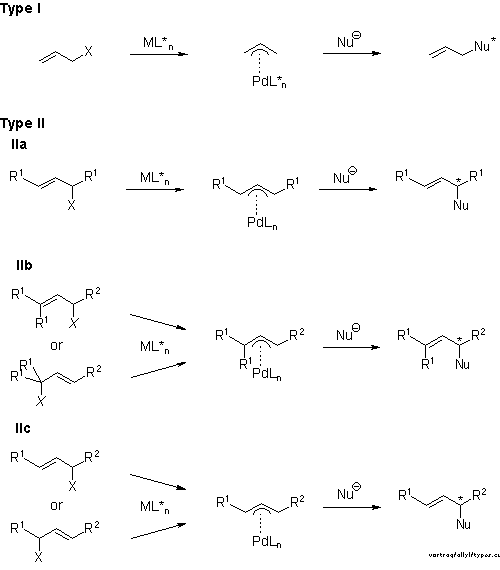
It is helpful for the understanding of substitutions at pi-allyl complexes to consider the fundamental types of substrates leading to chiral products given in Scheme 2. In reactions of type I a new chirality center is created in the nucleophile. The starting materials are achiral. In reactions of type II chiral racemic allylic derivatives are used as starting materials. The three subclasses are distinguished according to the possible modes of symmetry and isomerizations, via the well established[3] pi-sigma-pi-mechanism, of pi-allyl intermediates containing only achiral ligands at Pd.
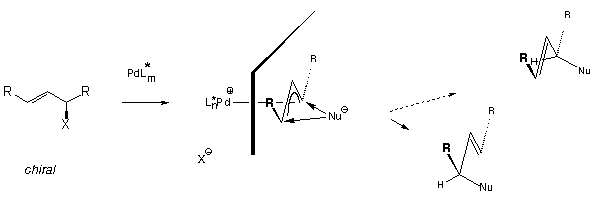
In this article we will particularly deal with reactions of type IIa which are more closely examined in Scheme 3. The reaction of the chiral, racemic allylic derivative with a Pd0 fragment yields the complex of a symmetric allylic cation. With achiral auxiliary ligands the intermediary pi-complex would be an achiral meso structure with enantiotopic electrophilic carbon atoms. The attack of a nucleophile would yield enantiomers in 1:1 ratio. In the presence of a chiral ligand L* terminal carbon atoms are diastereotopic, and hence enantiomers must be produced in unequal amounts.
One quite obvious problem associated with this reaction is the long distance between the chiral information provided by L* and the reaction path of the nucleophile. It was, therefore, believed for some time, that the (now) traditional C2-symmetric chelate ligands are not suited for differentiation of these two carbon atoms; indeed with diphosphines that gave excellent results in hydogenations, i.e. CHIRAPHOS, BINAP etc., results were not satisfactory, particularly with cyclic allylic substrates. However, over the last few years it was clearly demonstrated by Pfaltz with bisoxazolines[4] and Trost with diphosphines[5] that very high degrees of enantioselection are possible if a proper combination of substrate and ligand is chosen.
The transmittance of chiral information is more closely apparent from Scheme 4.

The combination of a C2-symmetric and a mirror symmetric object leads to an asymmetric object, as is nicely apparent for the bisoxazoline system: with respect to the coordination plane the pair of substituents R and R' are in a cis and in a trans relationship. Interaction of the cis-R/R' groups leads to distortion, in particular to weakening of the adjacent Pd-C bond as was demonstrated by x-ray crystal structures [4c]. Note that this effect occurs on the wings of the allylic system. Guidance of the nucleophile by hydrogen bonding is a concept successfully realized by the Ito-Hayashi team of Kyoto with their well-known phosphinoferrocenes that are particularly effective in allylic aminations[6]. Yet another concept is electronic differentiation that was realized by Faller for a stoichiometric substitution at the molybdenum complex shown in the last box of Scheme 4[7]. Here we have a nitrosyl and a carbonyl ligand of almost identical size and yet the reaction occurs exclusively cis to the better pi-acceptor NO.

A catalytic version of electronic differentiation was apparently first probed by Caesarotti with the ligand ProNOP with two slightly, by bonding to O or N, differentiated P atoms[8]. A fairly low level of enantioselectivity was achieved. We felt that a more pronounced difference in electronic as well as steric properties was required and, therefore, chose a hard, N, and a soft, P, S or Se donor combination. Realization of this proposal made use of the proven stability, variability and usefulness of the oxazoline moiety[9]. Aryl substituents were preferred as substituents at P because triarylphosphines are normally stable to air. The same concept was independently pursued by the groups of Pfaltz[10] and Williams[11]. With a different P-N chelate ligand, QUINAP, the group of J.M. Brown has also carried out allylic substitutions[12].