n-Butyl lithium is hexameric in the solid state[1] and in cyclohexane solutions. Why? Here I try to find out some of its secrets.
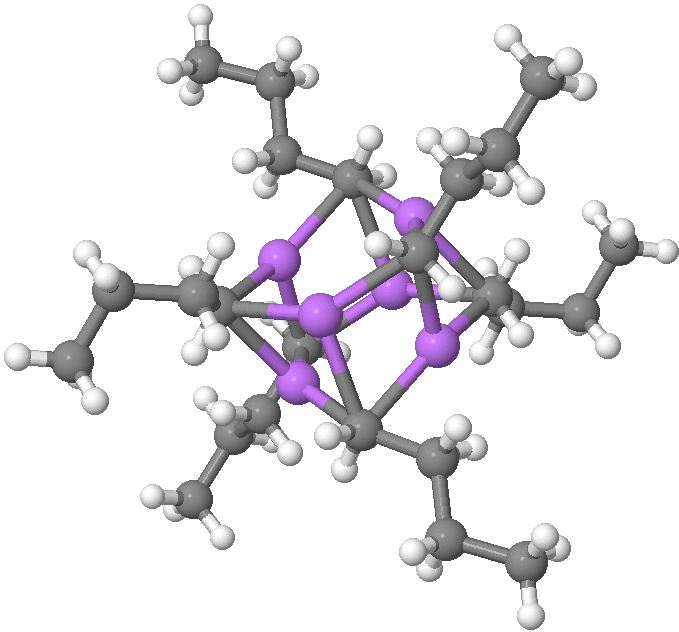
SUHBEC. CLICK FOR 3D.
The crystal structure reveals the following points of interest:
- Six lithium atoms form a cluster with triangular faces.
- An off-centre carbanion caps a triangular lithium face.
- Four of the butyl groups are in a fully extended antiperiplanar conformation
- But two di-axial n-butyl exhibit a gauche conformation.
The lithium cluster has twelve electrons available for bonding; if the Li is considered as Li+, balanced by six C– carbanions, the twelve electrons come from the six carbon lone pairs pointing towards each of six triangular faces. An ELF analysis can help identify how these twelve electrons are arranged. Shown below is the environment of a single Li-face, with the ELF basin ringed. It integrates to 2.08 electrons. So each tetrahedral cluster of three lithiums and one carbanion could be considered as a two-electron-four-centre bond, perhaps a natural progression from the two-electron-three-centre bonding found in a slightly less electron deficient system such as diborane.
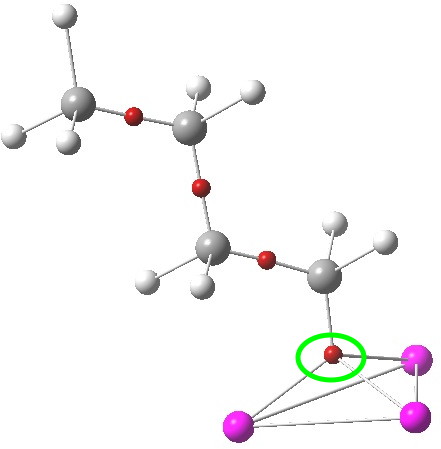
ELF basins. Click for 3D
NBOs (natural bond orbitals) reflect this character. An NBO represents a localised two-electron orbital, and analysis indeed reveals six such orbitals, each having the form shown below.
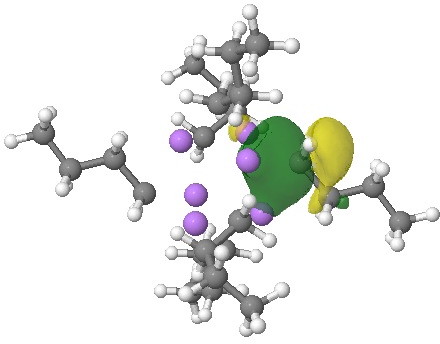
NBO. Click for 3D.
This picture in turn leads us to identify this system as spherically aromatic[2]. The three-dimensional equivalent of the Hückel rule is that any system with 2(N+1)2 σ or π electrons (or both) in a cluster can be considered aromatic/diatropic. In this case, N=0 and hence the magic count is 2 for each of the six CLi3 tetrahedra. The diatropic ring current might be manifested in the computed 1H NMR chemical shifts of the CH2– protons (-0.8ppm). Aromaticity does not immediately spring to mind with the name n-butyl lithium, but this unprepossessing molecule has six aromatic regions!
Each lithium atom is in turn hemispherically surrounded by three of these 2.08 electron basins (below, although the ELF centroid is very much biased towards the carbon, indicating considerable ionicity). What wonderful electronic economy! Despite there being only twelve electrons to be shared amongst six lithium atoms, each lithium manages nevertheless to surround itself with 6.24 electrons. All crammed into one half sphere, leaving a nice coordination hole; n-butyl lithium is after all a highly reactive species (even as a hexamer).
I want to finish by exploring the observation that two of the six n-butyl groups adopt a gauche conformation. In free n-butane itself, around 31% of the population adopts this shape, which curiously is around the same proportion as is found in the hexameric structure of n-butyl lithium. More generally, a search of the Cambridge database for compounds containing such groups reveals the following distribution; about 1 in 7.
Well, when you deprive a molecule of electrons, as any species with lithium must invariably suffer from, it is wonderful how the system responds. In this sense, a hexameric structure seems a very natural outcome. And it has brought us the two-electron-four-centre bond and the associated spherical aromaticity, both of which are a nice bonus.
Author
References
- T. Kottke, and D. Stalke, "Structures of Classical Reagents in Chemical Synthesis: (<i>n</i>BuLi)<sub>6</sub>, (<i>t</i>BuLi)<sub>4</sub>, and the Metastable (<i>t</i>BuLi · Et<sub>2</sub>O)<sub>2</sub>", Angewandte Chemie International Edition in English, vol. 32, pp. 580-582, 1993. https://doi.org/10.1002/anie.199305801
- A. Hirsch, Z. Chen, and H. Jiao, "Spherical Aromaticity inIh Symmetrical Fullerenes: The 2(N+1)2 Rule", Angewandte Chemie, vol. 39, pp. 3915-3917, 2000. https://doi.org/10.1002/1521-3773(20001103)39:21<3915::aid-anie3915>3.0.co;2-o
Tags: Cambridge, chemical shifts, conformational analysis, cyclohexane solutions, hexameric, spherical aromaticity, Tutorial material
Henry, Very interesting, as always, but NICS is conspicuously missing from your evidence. Also, I wonder about the simpler tetrahedral methyl lithium tetramer example, (CH3Li)4. Computed NICS on D3h Li3(+), Td Li4(2+), D6h Li6, etc. indicate very little induced diatropicty (much less than H3(+), Td H4(2+), D6h H6 and other “hydrogen aromatic surrogates.” However, we used NICS very early to study lithium clusters and found a few 3D “aromatic” ones, but this was never written up and published.
NICS was missing for several reasons.
1. I could not find a unique point to place it (the tetrahedra are not regular)
2. For small rings, I have always found NICS problematic; a NICS(0) probe picks up a lot of effects due to the proximity of nuclei, and this gets worse as the rings gets smaller. Perhaps there is a way of dissecting into NICSzz for this system, and if so it might be tried, but my experience with orienting 3D systems for this purpose is limited.
3. I suspect other ways of probing diatropic/paratropic ring currents should be also tried. Raphael Berger’s report of anapolar ring currents for a very “3D” helical system illustrates the difficulties that can manifest in such an approach however (see also 10.5560/ZNB.2012-0189).
My intention was perhaps to provoke people into seeing how well “3D aromaticity” in this cluster of 6 Li atoms stands up to close scrutiny.
1. n-butyllithium is a tetramer (like methyllithiim tetramer) in 1M THF, but it also is a hexamer in benzene, as in the solid state. Dimers or even monomers are present in other solvents, e.g., TMEDA
2. A well defined NICS point is the geometric center of the lithiums, four or six, as the case may be. QTAIM cage critical points (if they exist) may be used as well. But it makes little difference as can be shown easily by using a bunch of NICS points, grids or scans. These are more informative than single points.
3. How to overcome the problem of local shielding contaminations of primitive isotropic NICS(0) values, especially in small rings, is well established. Dissected NICS using appropriate tensor data for selected CMO’s or LMO’s works fine. Org Lett, 2006, 8, 863 and its SI give detailed instructions,
I wonder if the simpler model of thinking of this as simply ion pairing – a formal +1 charge ion each lithium and a formal -1 charge on C1 fo each butyl chain doesn;t explain the structure and reactivity just fine.
I am hesitant to argue any aromaticity here where the electron density between the C and Li is very small, and I expect the electron density at highlighted ELF basin is likely really small.
Just what property does “aromaticity” help to describe in this system?
In reply to Steve, the electron density for two electrons distributed amongst four centres would certainly be lower than that for eg a two-electron-two-centre “single” bond. The NBO two-electron localised wavefunction appears to confirm that it encompasses all four centres (and is different in nature from two-electron two-centre single bonds).
As Paul suggests with his comment ” a bunch of NICS points, grids or scans” one would really have to look at the current density across the whole (~ tetrahedral) region rather than try to identify just one single point for inspection.
As to what property does the designation “aromaticity” help describe, again I hinted at the measurable, ie the 1H chemical shift of the CH2 protons. Is the predicted high field shift due simply to adjacency to a localised carbanion, or would it be modified in any way by the diatropic ring currents expected of an “aromatic” system? I do not know the answer to that one; but I think it might be worth trying to get an answer to that question! I am here presuming that aromaticity nowadays can be best pinned down (it is a difficult concept to pin down indeed) by analysis of the magnetic properties of the system, rather than other metrics such as bond lengths or resonance energies.
Prompted by the questions posed by both Paul and Steve, I calculated the value of the nucleus independent chemical shift (NICS). There are many locations one could place it, but I decided to put it at the centroid of the ELF basins which I discussed in the post. These are approximately the centroid of the one carbon and three lithium atoms constituting the suggested 3D aromatic cluster. In this position, each of the six locations come out with a value of ~ -18.6 ppm (the zz tensor component of this is ~ -20.6). Such a value is normally taken as indicating significant diatropicity. The value at the centroid of the Li6 cluster is ~0 ppm.
I think this suggests that further analysis of this unusual form of apparent 3D aromaticity is probably worthwhile.
Steve raised the value of the electron density ρ, so I thought I would quantify it with a QTAIM calculation.
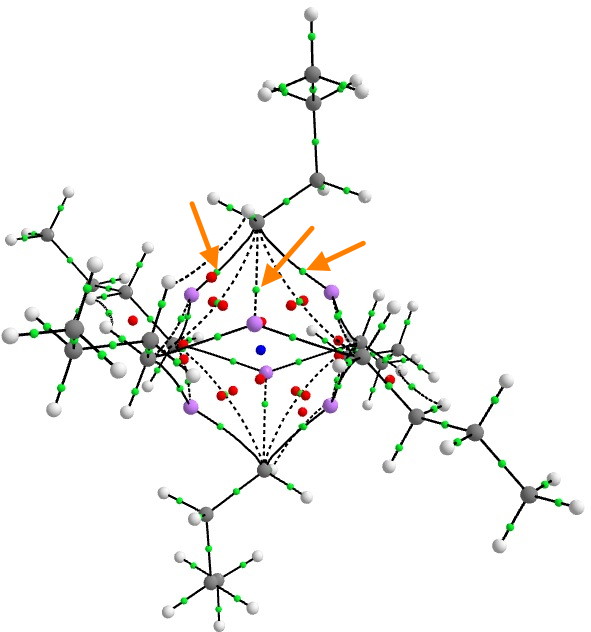
There are three two-centre bond paths (indicated with orange arrows) between any one carbanionic carbon and each of the three lithium atoms of any of the six tetrahedral clusters, for which the value of ρ is 0.026au at the bond-critical point. This is about 9 times less than the value for eg a C-C single bond. Bearing in mind these C-Li points all arise from just two electrons, the density, whilst low, is not unreasonably so. The BCP coordinates are located closer to the Li than the C, which indicates that the Li is the more electropositive of the two (no surprise there).
To put ρ into context, a strong hydrogen bond has a value of about 0.01 to 0.015, and a weak (dispersion) interaction has ~0.005 au. And finally, these are two-centre paths. The ELF analysis reveals the rather different picture of a two-electron-four-centre bond rather than localised two-centre interactions, which implies that this “bond” is stronger than the individual C-Li interactions seem to imply.
At any rate, I fancy few would imagine that an apparently simple metal alkyl compound could have such complex depths.
I was curious about the value of the density within the “aromatic” ring. I have published way back when a number of papers on the C-Li bond and its small electron density at the bond critical point. Just what is the density in the neighborhood of the ELF point that you have highlights? It will even smaller than the small value at the C-Li critical point – and so just what do you want to make of all of this very small density?
As a follow up, I include here some details of a calculation on Li4(2+). It is tetrahedral with Li…Li distances of 3.5Å. A NICS probe at the centroid has the diatropic value of -10.9 pm (ωB97XD/6-311G(d,p) calculation), coinciding with an ELF centroid for a basin integrating to 1.93e. The QTAIM is different however (below). A non-nuclear attractor is located at the centroid, with four bond paths to it from each of the Li atoms. This really does merit the description of a two-electron-four-centre interaction.
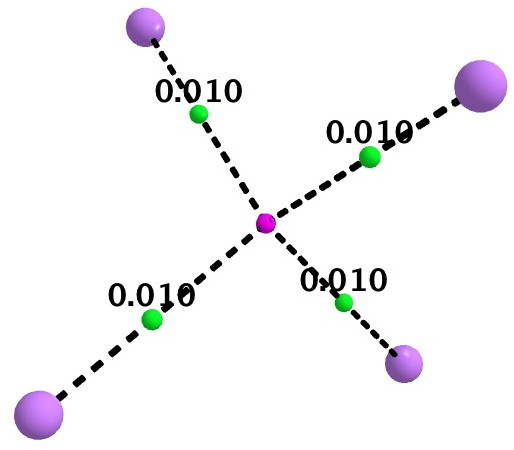
If Li4(2+) represents the ideal of a homonuclear “3D aromatic” cluster, the n-butyl lithium system may be closer to an ionic heteronuclear cluster, in the limit tending towards an entirely ionic, non-interacting ion-pair. n-butyl lithium is on the slider somewhere between these two extremes!
In reply to Steve, the ELF centroid does not coincide with an critical point in the QTAIM. So I will have to ponder how to inject the coordinates of the former into the latter analysis so as to extract a density at that point.
What do I make of all this small density? Well, only two electrons occupy this region, so any single pair of nuclei will invariably show a low density. Should we call any of this a bond? Certainly, for any given two nuclei, any “bond” between them is obviously similar to (perhaps slightly stronger than) a hydrogen bond. That is certainly called a bond! But if one regards the bonding as a two-electron-four-centre bond, then there is only one bond really, but it encompasses four nuclei. A density at any single point is then probably not particularly helpful.
I think we might end up arguing about how many angels can dance on the point of a needle. What is a polar bond vs what is an electrostatic attraction between two ions. As I note in the comment above this one in regard to Li4(2+), we probably can agree that n-butyl lithium is somewhere between a pure ion pair (whatever that is) and a pure two-electron-four-centre cluster. Quite where along that slider it is will probably depend on your context.
I computed a coarse grid of NICS to see what happens around the vicinity of the (CH3Li)6 hexamer. Truly aromatic systems should display negative NICS values inside but positive NICS values outside of the cage/(or ring). But this does not happen for (CH3Li)6! See two views below (the largest isotropic NICS value around the C atom is –48.5 ppm, cage center: –1.05 ppm, Li3 three membered ring center: –4.58 ppm). It seems like any “diatropicity” inherent to (CH3Li)6 arises predominately from local circulations around the highly shielded carbanion C centers. On this basis, it is probably difficult to justify (CH3Li)6 as being aromatic, despite its negative NICS values at certain positions in the molecule.
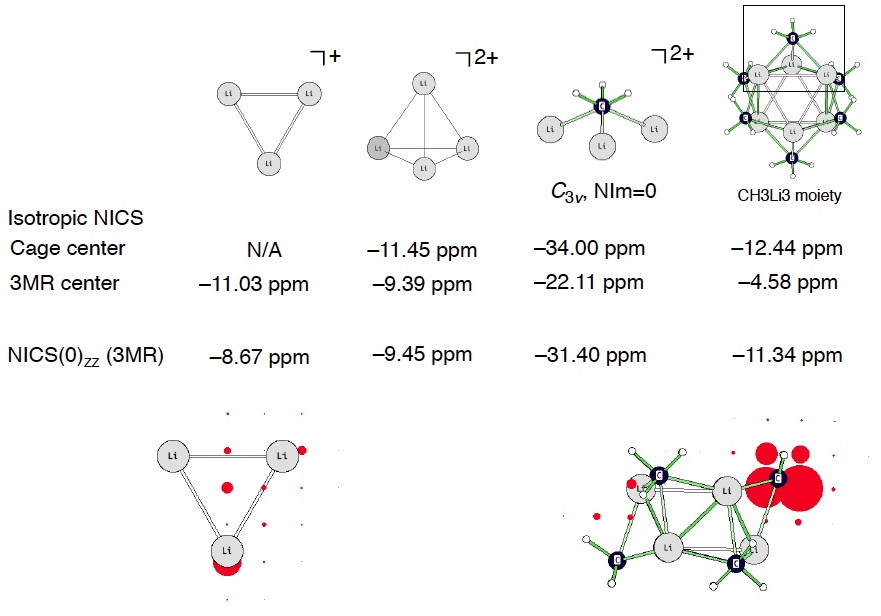
I also checked the 1H chemical shifts of the CH3 protons in (CH3Li)6 vs. the methyl anion, CH3(–). The protons in (CH3Li)6 are indeed downfield relative to CH3(–) (supportive of possible aromaticity), but only by 0.22 to 0.50 ppm!
The currents look like those of an aggregate of alternating anions and cations. Overall currents are close to zero but all in all diamagnetic.
Thanks all for the various great insights.
They have helped make for a fascinating analysis of what might have appeared to be an insignificant molecule. I will point this discussion out to our students; I am sure they will love it!
PS: The currents in particular do not resemble cluster/3D aromatic currents, which are diatropic vortices passing through the hole cluster, without central paratropic vortices.
Can you please provide me the coordinate file for the Butyl lithium hexamer. I was trying to optimize it.
Coordinates can be obtained from any of the 3D models visualised by clicking on the image as per instruction and using the JSmol menu, or by clicking on any of the three hyperlinks in the text which lead to data on a data repository.