The benzidine rearrangement is claimed to be an example of the quite rare [5,5] sigmatropic migration[1], which is a ten-electron homologation of the very common [3,3] sigmatropic reaction (e.g. the Cope or Claisen). Some benzidine rearrangements are indeed thought to go through the [3,3] route[2]. The topic has been reviewed here[3].

In this post, I offer a calculated transition state and IRC for this reaction, to see what insights might accrue. How was this obtained?
- At the ωB97XD/6-311G(d,p)/SCRF=water level. This procedure would allow for any dispersion-like effects to be allowed for in the π-π-stacking.
- The rearrangement is normally promoted by acid, and the active species is thought to be diprotonated[4]‡ (although monoprotonated catalysis is also observed[1]. Here I report just the diprotonated route, together with chloride anions to balance the charges, and have added a continuum water field to allow this double ion-pair to be at least partially stabilised.
- The rate determining step is the N-N cleavage/C-C bond formation. This is followed by presumed rapid proton transfers, which are not modelled here.
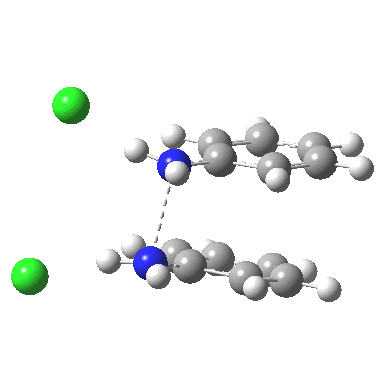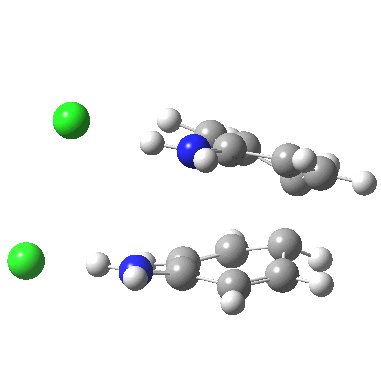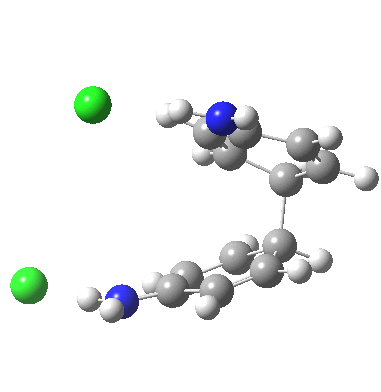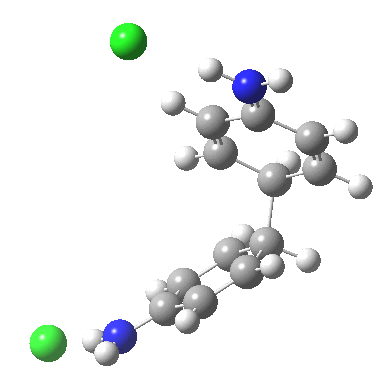
![The [5,5] transition state for the benzidine rearrangement. Click for 3D.](http://www.ch.imperial.ac.uk/rzepa/blog/wp-content/uploads/2013/01/benzidine-55.jpg) The [5,5] transition state for the benzidine rearrangement. Click for 3D. |
 |
 |
 |
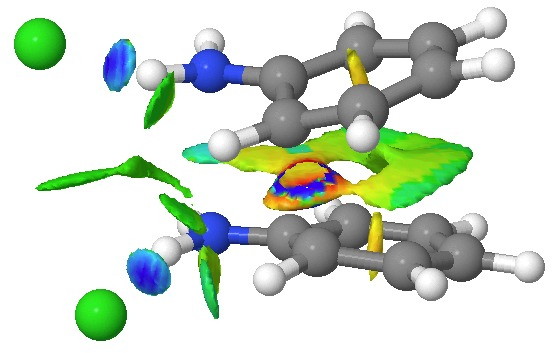
This [5,5] transition state is 2.9 kcal/mol lower in ΔG‡298 than the transition state for the isomeric [3,3] rearrangement. The NCI (non-covalent-interactions) shows the forming C-C bond to be on the border of covalent, and non-covalent (blue), but that the π-π-stacking region is all weakly attractive (green). You can also observe the strong hydrogen bonds between the chloride anion and an N-H group (blue), and the weak attractive zones between the two nitrogen centres, between the chloride and the ortho-C-H hydrogens, and even between the two chloride anions (blue-green or green). I should point out that the initial position for these anions was over the aryl ring, but they migrated to the NH region during optimisation of the transition state.
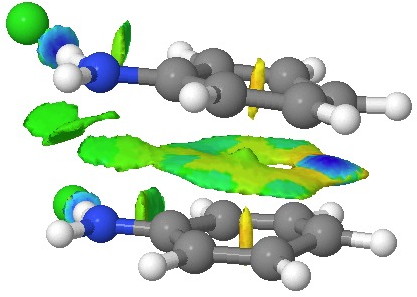
NCI surface. Click for 3D.
The molecular electrostatic potential (isosurface = 0.11 au) shows both aryl rings as a single unit attracted by a positive potential (blue)
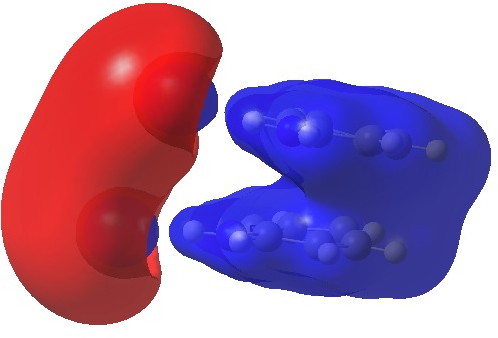
Calculated electrostatic potential. Click for 3D.
The highest-occupied molecular orbital shows the two bonds involved in the [5,5] shift (N-N and C-C) are both bonding, but more significantly, the central region of the two stacked aryl rings is also bonding. This is a clear manifestation of a π-complex, which the benzidine rearrangement has often (and it has to be said controversially) described as, and which elevates this particular reaction from that of a simple bond forming/bond cleaving sigmatropic. Another way of looking at it is that secondary orbital interactions (such as often invoked in Diels-Alder cycloadditions) are exceptionally important here.
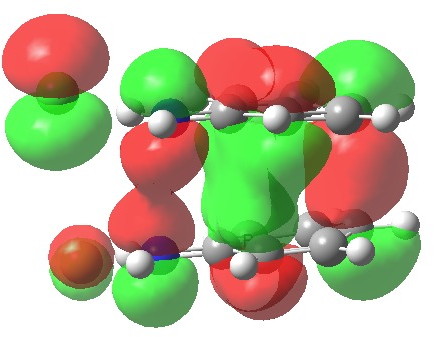
HOMO for 5,5 benzidine rearrangement. Click for 3D.
The LUMO is strongly antibonding in that region; indeed adding two electrons to form a 12-electron process would be strongly destabilising. In this regard, this unusual sigmatropic reaction follows the same 4n+2 electron rule as more conventional ones.
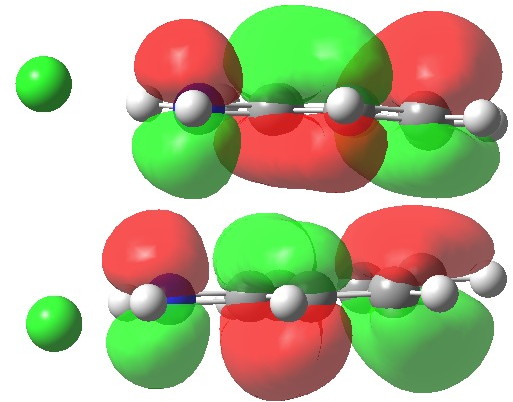
LUMO. Click for 3D.
The next two diagrams illustrate the competing (higher energy) [3,3] shift, which also has some π-complex character.
![A [3,3] alternative to the benzidine rearrangement. Click for 3D.](http://www.ch.imperial.ac.uk/rzepa/blog/wp-content/uploads/2013/01/benzidine-33.jpg)
A [3,3] alternative to the benzidine rearrangement. Click for 3D.
NCI surface for 3,3 rearrangement. Click for 3D.
I will end with three autobiographical notes.
- The benzidine rearrangement was one of the earliest reactions I did in my home laboratory, at the age of about 13. As I recollect, I prepared about 1.5 grams (blissfully ignorant of how carcinogenic it is), and used it via diazotization to couple to phenol. My fascination with chemistry most certainly started with colour (and how to express the bonding in nitric oxide).
- About eight years later, I was about to commence my Ph.D. studies. The objective was to use kinetic isotope effects to infer the structure of transition states. In my case (proton transfers to indoles) I never did achieve this objective. But it is noteworthy that the mechanism of the benzidine rearrangement was largely unravelled using such isotopic studies.
- By 1974 as a post-doctoral researcher, I had moved on to studying mechanisms using quantum theory and had decided that it was easier to invert the use of isotope effects by predicting a transition structure using this method, and then seeing if the computed isotope effects matched the experiment. We did this for the Diels-Alder reaction[5] and more generally[6], and then for some gas-phase eliminations[7], this latter being my first entirely independent publication.
- So, putting all this together, one might infer that armed with a computed transition state structure for the benzidine rearrangement, it is trivial to compute the kinetic isotope effects and hence to see if they correspond to those measured. You might expect a report on this in a future post here.
‡ Crystal structures of diprotonated dimethyl hydrazines[4] show a N-N bond length of ~1.45Å (typical counter-anions being nitrate, perchlorate or sulfate). That calculated for the diprotonated diphenyl hydrazine is ~2.5Å, which suggests that with the phenyl group, electrons from the N-N region may be borrowed to contribute to the π-π-complex.
Author
References
- H.J. Shine, K.H. Park, M.L. Brownawell, and J. San Filippo, "Benzidine rearrangements. 19. The concerted nature of the one-proton rearrangement of 2,2'-dimethoxyhydrazobenzene", Journal of the American Chemical Society, vol. 106, pp. 7077-7082, 1984. https://doi.org/10.1021/ja00335a035
- H.J. Shine, L. Kupczyk-Subotkowska, and W. Subotkowski, "Heavy-atom kinetic isotope effects in the acid-catalyzed rearrangement of N-2-naphthyl-N'-phenylhydrazine. Rearrangement is shown to be a concerted process", Journal of the American Chemical Society, vol. 107, pp. 6674-6678, 1985. https://doi.org/10.1021/ja00309a041
- H.J. Shine, "Reflections on the π‐complex theory of benzidine rearrangements", Journal of Physical Organic Chemistry, vol. 2, pp. 491-506, 1989. https://doi.org/10.1002/poc.610020702
- C.M. Sabaté, and H. Delalu, "Energetic Salts of Symmetrical Dimethylhydrazine (SDMH)", European Journal of Inorganic Chemistry, vol. 2012, pp. 866-877, 2011. https://doi.org/10.1002/ejic.201101115
- M.J.S. Dewar, S. Olivella, and H.S. Rzepa, "Ground states of molecules. 49. MINDO/3 study of the retro-Diels-Alder reaction of cyclohexene", Journal of the American Chemical Society, vol. 100, pp. 5650-5659, 1978. https://doi.org/10.1021/ja00486a013
- S.B. Brown, M.J.S. Dewar, G.P. Ford, D.J. Nelson, and H.S. Rzepa, "Ground states of molecules. 51. MNDO (modified neglect of diatomic overlap) calculations of kinetic isotope effects", Journal of the American Chemical Society, vol. 100, pp. 7832-7836, 1978. https://doi.org/10.1021/ja00493a008
- H.S. Rzepa, "MNDO SCF-MO calculations of kinetic isotope effects for dehydrochlorination reactions of chloroalkanes", Journal of the Chemical Society, Chemical Communications, pp. 939, 1981. https://doi.org/10.1039/c39810000939
Tags: higher energy, Historical, π-complex, pericyclic, Reaction Mechanism
[…] Chemistry with a twist « The mechanism of the Benzidine rearrangement. […]
[…] The match between experiment and theory for TS(CC) is reasonable (given the approximations in both the theory and the difficulty of the experiments and ensuring isotopic purities) but not so for TS(NN). But TS(CC) is a “stepwise-concerted” reaction as a closed shell singlet; as shown in the IRC computed from TS(CC. […]
[…] noticed my noting in previous posts that the benzidine rearrangement proper is normally promoted by double protonation, and that reaction via monoprotonation has a significantly higher barrier. So what are the […]
[…] few posts back, I explored the “benzidine rearrangement” of diphenyl hydrazine. This reaction requires diprotonation to proceed readily, but we then […]