A few years back, I took a look at the valence-shell electron pair repulsion approach to the geometry of chlorine trifluoride, ClF3 using so-called ELF basins to locate centroids for both the covalent F-Cl bond electrons and the chlorine lone-pair electrons. Whereas the original VSEPR theory talks about five “electron pairs” totalling an octet-busting ten electrons surrounding chlorine, the electron density-based ELF approach located only ~6.8e surrounding the central chlorine and no “octet-busting”. The remaining electrons occupied fluorine lone pairs rather than the shared Cl-F regions. Here I take a look at ClMe3, as induced by the analysis of SeMe6.
The difference between ClF3 and ClMe3 is that octet-excess electrons (two in this case) in the former can relocate into fluorine lone pairs by occupying in effect anti-bonding orbitals and hence end up not contributing to the central atom valence shell.‡ With ClMe3 the methyl groups cannot apparently sustain such lone pairs, at least not distinct from the Cl-C bond region. So might we get an octet-busting example with this molecule? A ClMe3 calculation (ωb97xd/6-311++g(d,p)) reveals a molecule with all real vibrational modes (i.e. a minimum, FAIR data DOI: 10.14469/hpc/3241) and ELF (FAIR data DOI 10.14469/hpc/3242)† basins as shown below:
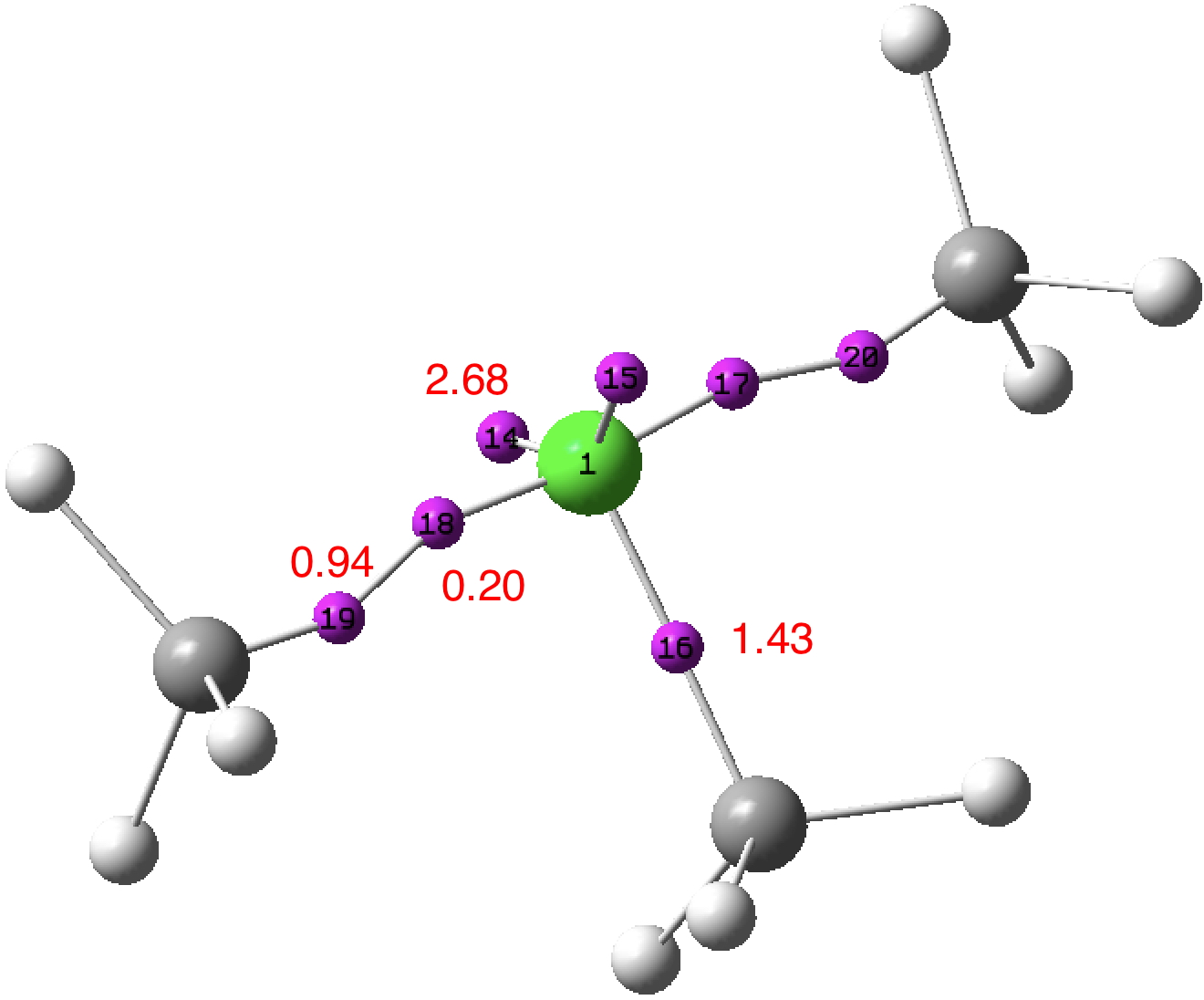
Density-derived approach: Two of the C-Cl bonds each exhibit two ELF basins; one disynaptic basin (0.94e) and one monosynaptic basin (0.20e) closer to the chlorine. The former pair integrate to 1.88e, density which largely arises from carbon (natural charge -0.84) and which contribute to a total integration for these carbons of 7.17e. The latter pair contributes to a total chlorine integration of 7.19e. The angle subtended at chlorine for the two 2.68e “lone pair” basins is 141°. Thus an inner, octet-compliant, valence-shell for chlorine is revealed, plus an expanded-octet outer one into which the two additional electrons go. The latter contribute to forming an octet-compliant carbon valence shell, but may be considered as not contributing to the valence shell of the other atom of the pair, the chlorine. An endo lone-pair rather than the more usual exo lone-pair if you will. These results reveal that the molecular feature we know as a (single) “bond” may in fact have more complex inner structures or zones, something we do not normally consider bonds as having. In this model, these zones are not invariably considered as shared between both the atoms comprising the bond.
Orbital-derived approach: NBO analysis (FAIR data DOI: 10.14469/hpc/3241) reveals the chlorine electronic configuration as [core]3S(1.83)3p(4.67)4S(0.01)3d(0.03)5p(0.02,) showing very little population of the Rydberg shells (4s, 3d, 5p) occurs (0.13e in total). This method of partitioning the electrons allocates a chlorine Wiberg bond index of 2.00 and the methyl carbon bond index of 3.83. If the regular valence of Cl is taken as 1, then the central chlorine can be regarded as non-Rydberg hypervalent (the electrons in the 0.94e basins are taken as contributing to the chlorine bond index).
The carbon-halogen bond internal structures simplify for Br (DOI: 10.14469/hpc/3248, 10.14469/hpc/3250) and I (DOI: 10.14469/hpc/3249, 10.14469/hpc/3247); for each only a single ELF basin is located and the NBO Br and I bond indices are respectively 2.10 and 2.1. This is not due to incursion of Rydberg hypervalence (Br: [core]4S(1.83)4p(4.46)5S(0.02)4d(0.03)6p( 0.01); I: [core]5S(1.82)5p(4.29)6S(0.02)5d(0.02)6p(0.01) ) but of a merging of the carbon and halogen valence basin such that the ELF contributions to each cannot be deconvoluted. In each case the NBO bond indices of ~2 suggest hypervalency for the halogen.
What have we learnt? That the shared electron (covalent) bond can have complex internal features, such as two discrete basins for the apparently shared electrons. How one partitions these electrons can influence the value one obtains for the total shared electron count and hence whether the octet is retained or expanded for main group elements such as the halogens. And finally, that hypervalence and hyper-coordination are related in the orbital model at least. Thus along the series MenI (n= coordination number 1,3,5,7), the values of the Wiberg bond index at the halogen progress as 1.0, 2.1, 3.1 (DOI: 10.14469/hpc/3236) and 4.01 (DOI: 10.14469/hpc/3238), or one extra atom bond index per electron pair. Given this, it seems useful to retain the distinction between the terms hypervalence and hyper-coordination, but also recognize that we still may have much to learn about the former.
‡See the previous post on SeMe6 for a more detailed discussion.
† The FAIR Data accompanying this blog post is organised in a new way here. All the calculations are collected together with an over-arching DOI: 10.14469/hpc/3252 associated with this post, with individual entries accessible directly using the DOIs given above. The post itself has a DOI: 10.14469/hpc/3255 and the two identifiers are associated with each-other via their respective metadata. A set of standards (https://jats.nlm.nih.gov) with implementation guidelines for e.g. repositories, authors and publishers-editors are expected in the future to establish infra-structures for cross-linking narratives/stories with the data on which they are based.
Author
Tags: Chemical bond, chemical bonding, Chemistry, Chlorine, Covalent bond, Lone pair, Oxidizing agents, Quantum chemistry, Stereochemistry, Valence, VSEPR theory
Here is an ELF analysis of square pyramidal ClMe5 (FAIR data DOI: 10.14469/hpc/3257). It shows the strange bifurcated internal structure for two of the basal C-Cl bonds. It is an odd analysis, since the other two basal Cl-C bonds differ, for reasons I cannot rationalise.
One problem associated with the density-based ELF approach is connected with what might be described as inhomogeneities in the density due directly to the basis set. A basis set is constructed of overlapping gaussian functions, and not all basis sets have been necessarily constructed to be internally free of inhomogeneities. Thus the 6-311++G(d,p) takes the base 6-311 gaussian functions and overlays on these additional functions, both diffuse (++) and polarisation (d,p). The overall basis set therefore has not necessarily been optimised to remove any inhomogeneities. These in turn may lead to small oscillations, if you will, in the density along a bond, and these in turn may propagate to the ELF function. When this is used to localise and then integrate basins, you may get features in this process that are just artefacts of the basis set. Still, it would be interesting to see if basis sets optimised for ELF exist?
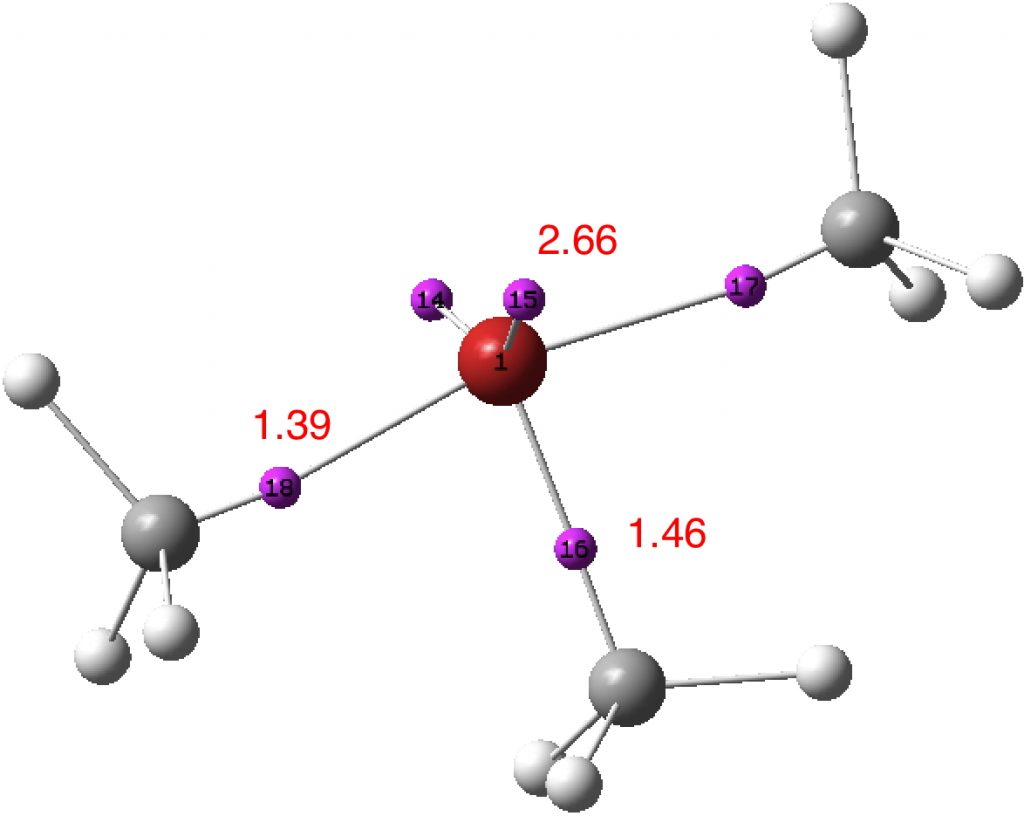
A first test of this was to re-evaluate the ELF analysis using a Def2-TZVPP basis, where all the polarisation and diffuse functions are optimised concurrently and not added in layers (a well-balanced basis set). The result is shown below (ran using a large grid of 600,600,600 to reduce integration artefacts). The splitting of the C-Cl valence basins is still apparent! So it may be a real phenomenon rather than something induced by the method used.
I would also add that such bond splitting can be seen in charge-shift bonds, such as e.g. F2.
An ELF analysis (ωB97XD/Def2-TZVPP) using the MultiWFN program (ultra high resolution option) gives an identical result.
The CASSCF(8,8) or CASSCF(12,12) multi-reference methods give very similar results:
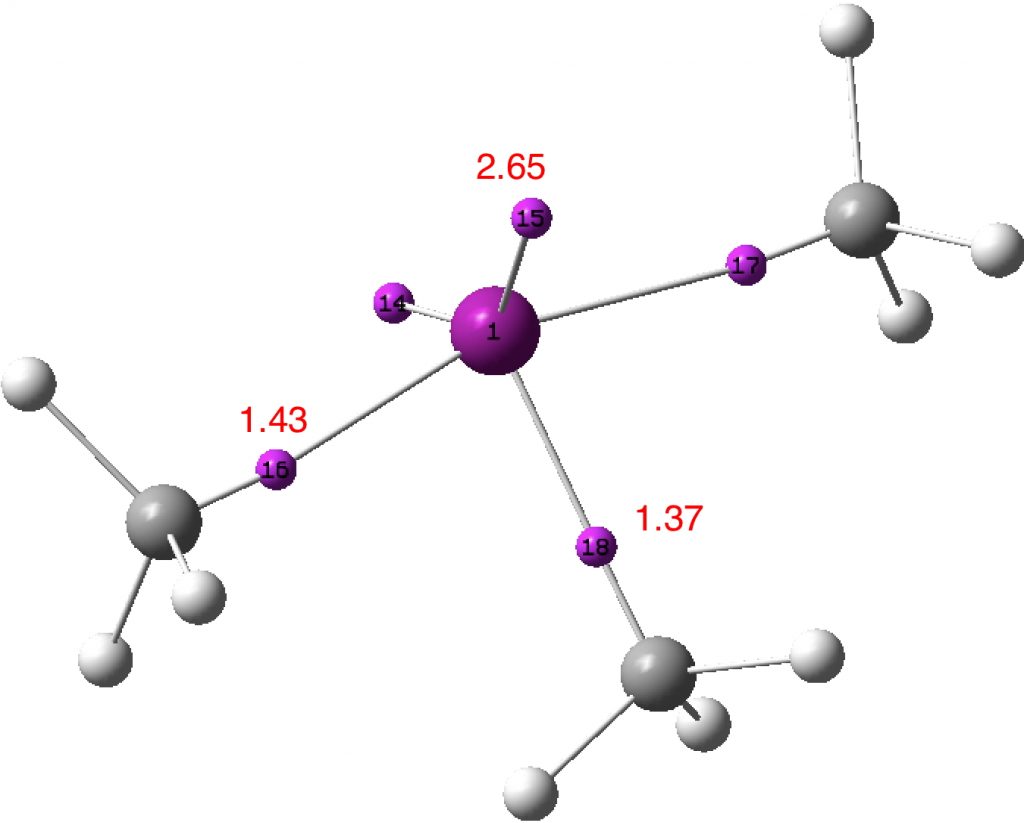
I have repeated the odd looking ELF analysis for ClMe5 using the Def2-TZVPP basis set (FAIR data doi: 10.14469/hpc/3282), with much more sensible looking results. Put simply, the four basal Cl-Me bonds all show the binary basin structure revealed in ClMe3. In effect, of the formally twelve electrons surrounding the chlorine in the valence shells (7 from Cl, 5 from the five Me groups), approximately 4 go into the basins closer to carbon and the remainder form the octet shell around Cl.
It is probably safest to use well-balanced basis sets for ELF rather than arbitrarily extended ones in future!
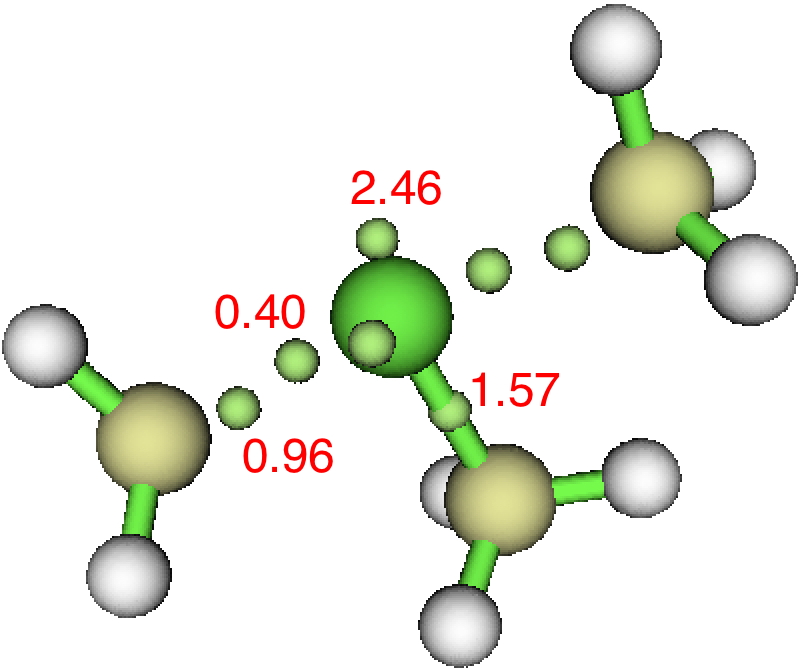
I noted in a comment above that ELF basin-splitting was also associated with F2. Here are two plots of the ELF attractors using respectively DFT (ωB97XD/Def2-TZVPPD, FAIR Data DOI: 10.14469/hpc/3294) and CASSCF(10,10)/Def2-TZVPPD (FAIR Data DOI: 10.14469/hpc/3295) showing similar splitting of the ELF basins along the axis of the F-F bond (along with circular attractors surrounding each atom and corresponding to the density from the lone pairs).
1. The multi-reference CAS function shows rather greater “splitting” of the F-F basins than the single-reference DFT method.
2. The total population of the two F-F covalent basins is low, at 0.26 – 0.12e.
3. This fits very nicely with the character of the F-F bond, which is described as a charge-shift bond. This means a far greater contribution from valence structures such as F–.F+, where the ELF function would show up in the monosynaptic F lone pairs, rather than the disynaptic covalent F:F form.
4. This implies that such charge-shifted bonds may have some significant multi-reference contributions.