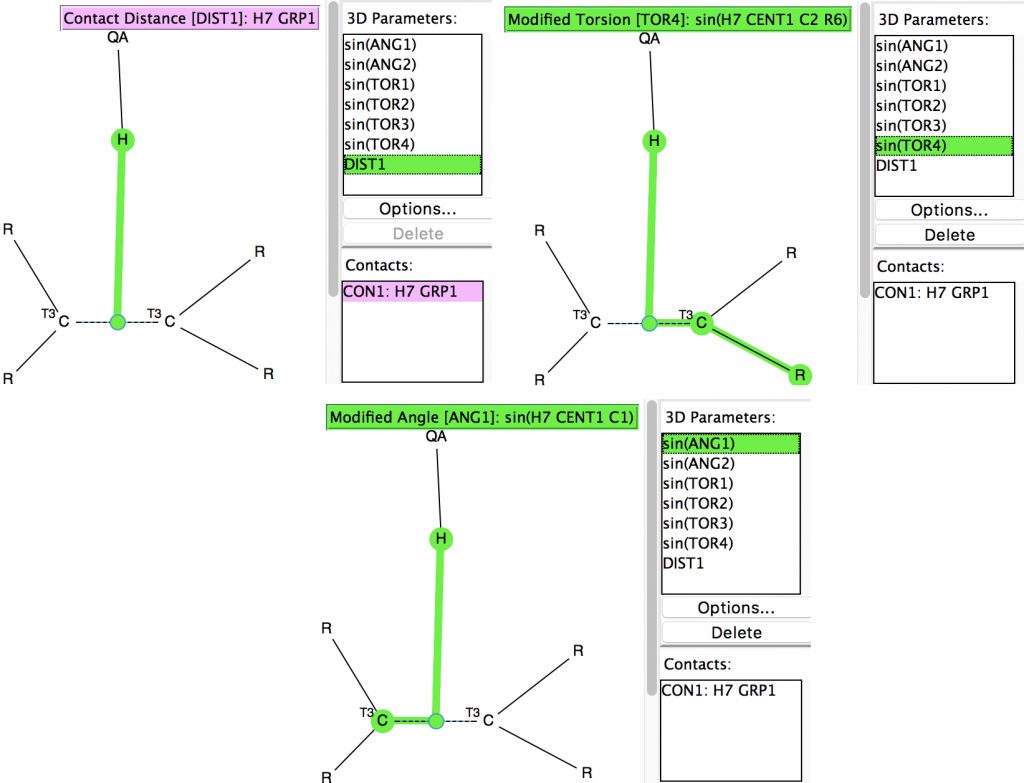
Back in the early 1990s, we first discovered the delights of searching crystal structures for unusual bonding features.[1] One of the first cases was a search for hydrogen bonds formed to the π-faces of alkenes and alkynes. In those days the CSD database of crystal structures was a lot smaller (<80,000 structures; it’s now ten times larger) and the search software less powerful. So here is an update.
The search query (dataDOI:10.14469/hpc/2473) is shown below:
- A mid-point (centroid) of a C-C bond (of any type) is defined, but the carbons are each restricted to being 3-coordinate, with the substituents R being either C or H.
- The distance to a hydrogen (attached to group QA, where QA is any one of N,O,F,Cl, i.e. acidic H) is defined.
- The properties of the alkene are defined by the sines of the two angles subtended at the centroid. This defines how perpendicular the QA-H hydrogen bond is to the C-C bond.
- Four torsions R-C-centroid-H are defined by their sines. The mean of the absolute values of these will define how orthogonal the approach of the hydrogen to the π-π plane is.
- Further constraints in the search are no disorder, no errors, R < 0.05, the H atom position is normalised and this position is defined as being <2.5Å from the C-C bond centroid, which is ~0.3Å < the sum of the van der Waals values for C and H.
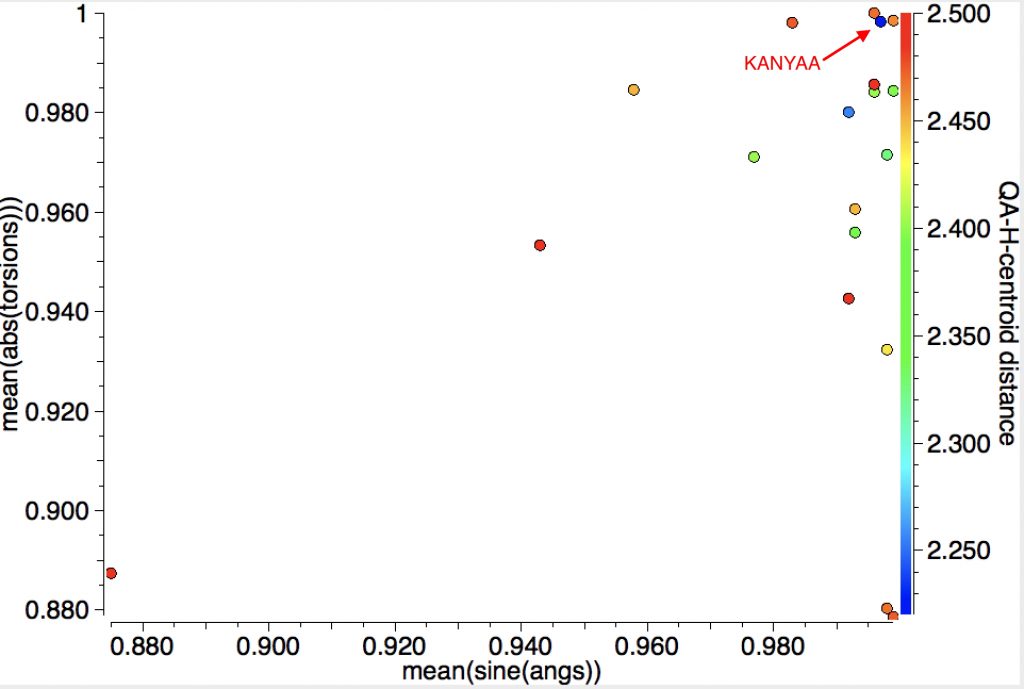
The first search is limited to intermolecular contacts between the C-C bond and the H and reveals that for most of the 18 hits, the H approach is close to perpendicular to the centroid but the inclination to the π-π plane is more scattered. The most interesting (shortest H…centroid contact of ~2.22Å, orthogonal approach) can be inspected as KANYAA (dataDOI: 10.5517/CC8JRQ7).
 When the search is repeated for intramolecular contacts, rather shorter distances are obtained for 88 hits and with more variation in the angles of approach. The most interesting candidate (blue dots) is IGELAJ[2] (dataDOI: 10.5517/CC14PBW1 ) which has the very short intramolecular H approach of 1.90Å to the C-C centroid corresponding to ~2.04Å to the carbons, a contraction of ~0.8Å from the van der Waals sum.
When the search is repeated for intramolecular contacts, rather shorter distances are obtained for 88 hits and with more variation in the angles of approach. The most interesting candidate (blue dots) is IGELAJ[2] (dataDOI: 10.5517/CC14PBW1 ) which has the very short intramolecular H approach of 1.90Å to the C-C centroid corresponding to ~2.04Å to the carbons, a contraction of ~0.8Å from the van der Waals sum.
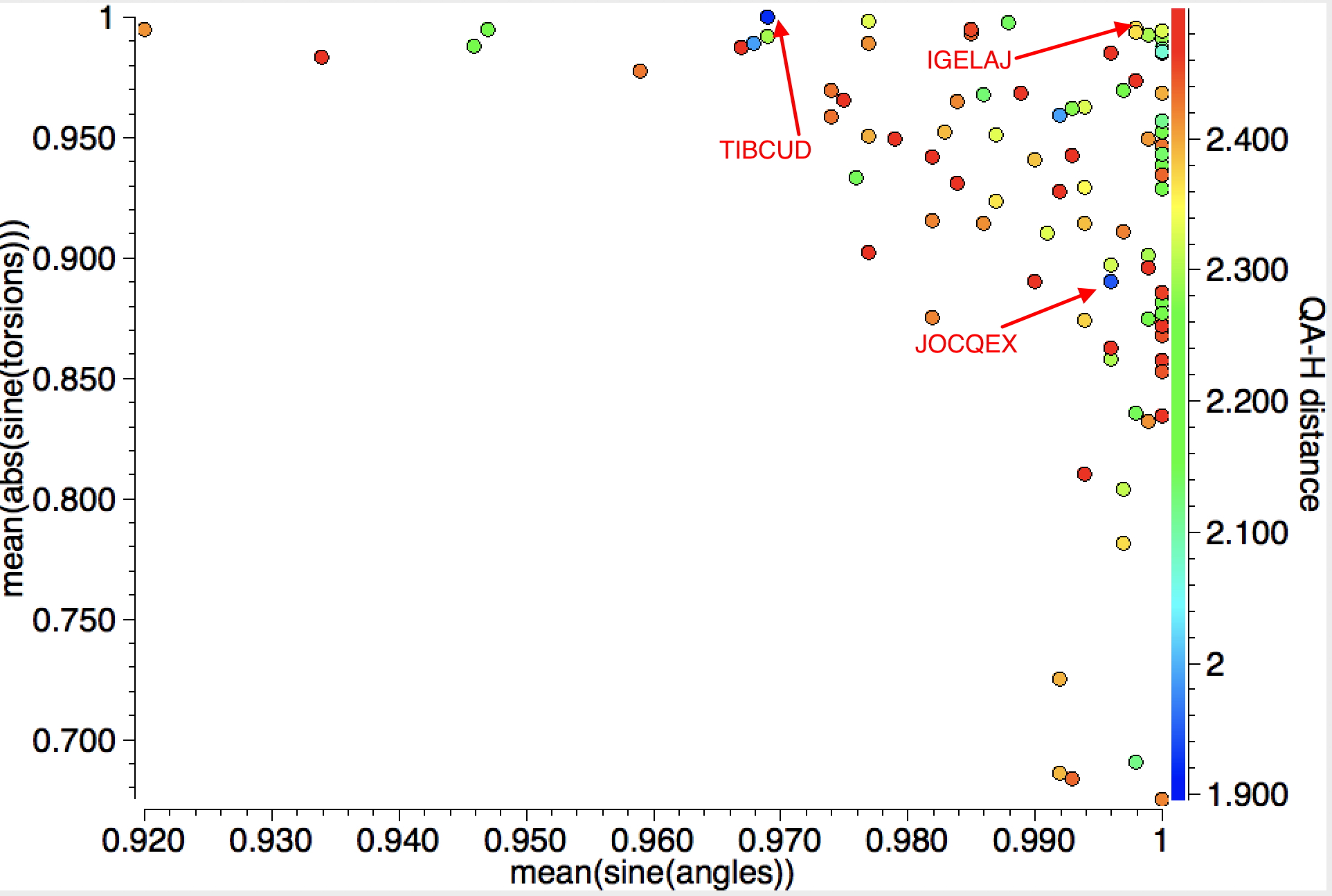
The authors remarked[2] “that it possesses a better defined intramolecular hydrogen bond compared to the usual molecules for which it is noted“. They also note JOCQEX, which is present in the above plot, but for which there is a non-orthogonal approach of the hydrogen bond to the π-π plane. The authors do not mention TIBCUD[3] (dataDOI: 10.5517/CCPL0FP), which has a similar close approach of 1.92Å to the C-C centroid, but at an angle inclined to the C-C axis.
IGELAJ, as an intramolecular H-bond, was amenable to calculation of its geometry and properties (inter-molecular interactions would ideally require the periodic lattice to be computed), with the observation[3] that “another test was to compare the energy calculation of IGELAJ to a non-hydrogen-bound version where the OH bond is rotated 180°” and “the results predict IGELAJ to be 7.30 kcal more stable than the non-hydrogen-bound version”. This value, if correct, is indeed typical of a very strong hydrogen bond!
Pedant (curious?) as I am, I wanted to be clear what kind of calculated energy was being reported. Was it the difference in total energies, or the energies corrected for ZPE (zero-point-energy) as ΔH or the free energies for which entropy is included as ΔG? The article[3] itself is unclear on this aspect and no energies are reported in the supporting information. This is an illustration that “supporting information” in most current incarnations may often not provide crucial information; only a full deposition as the management of research (RDM) of FAIR data can provide. This process is illustrated for my own calculations of this system (ωB97XD/Def2-TZVPP, dataDOIs: 10.14469/hpc/2474, 10.14469/hpc/2475), which reveals that ΔG298 4.8 kcal/mol and ν 3761 cm-1. In comparison when the OH bond is rotated 180° the wavenumber goes up 3956 cm-1, a difference of 195 cm-1 is calculated, which is indeed a large red-shift. But the “non-hydrogen-bound version where the OH bond is rotated 180°” is not a valid reference point for a non-hydrogen bonded isomer, since it manifests instead as a transition state for OH rotation with νi 166 cm-1, there being no minimum other than the π-facially hydrogen bonded one (dataDOI: 10.14469/hpc/2476). So, for the lack of a suitable reference system, we cannot conclude what the strength of this particular hydrogen bond is, nor make any conclusions about it being unusually strong.
So IGELAJ holds the current record for the shortest π-facial hydrogen bond to an alkene, but not necessarily the strongest! I wonder if this record might be broken with the aid of further computational design and prediction?
Author
References
- H.S. Rzepa, M.H. Smith, and M.L. Webb, "A crystallographic AM1 and PM3 SCF-MO investigation of strong OH ⋯π-alkene and alkyne hydrogen bonding interactions", J. Chem. Soc., Perkin Trans. 2, pp. 703-707, 1994. https://doi.org/10.1039/p29940000703
- M.D. Struble, M.G. Holl, G. Coombs, M.A. Siegler, and T. Lectka, "Synthesis of a Tight Intramolecular OH···Olefin Interaction, Probed by IR,<sup>1</sup>H NMR, and Quantum Chemistry", The Journal of Organic Chemistry, vol. 80, pp. 4803-4807, 2015. https://doi.org/10.1021/acs.joc.5b00470
- B. Ndjakou Lenta, K.P. Devkota, B. Neumann, E. Tsamo, and N. Sewald, "4-(1,1-Dimethylprop-2-enyl)-1,3,5-trihydroxy-2-(3-methylbut-2-enyl)-9<i>H</i>-xanthen-9-one", Acta Crystallographica Section E Structure Reports Online, vol. 63, pp. o1629-o1631, 2007. https://doi.org/10.1107/s1600536807009907
Tags: calculated energy, chemical bonding, Chemistry, Crystal, crystallography, energy, energy calculation, Intermolecular forces, Nature, search query, search software, Supramolecular chemistry
In the search above, I restricted myself to “acidic” hydrogen attached to electronegative elements interacting with a C=C π-bond. Here I extend things by also including hydrogens attached to carbon or boron for an intramolecular search.
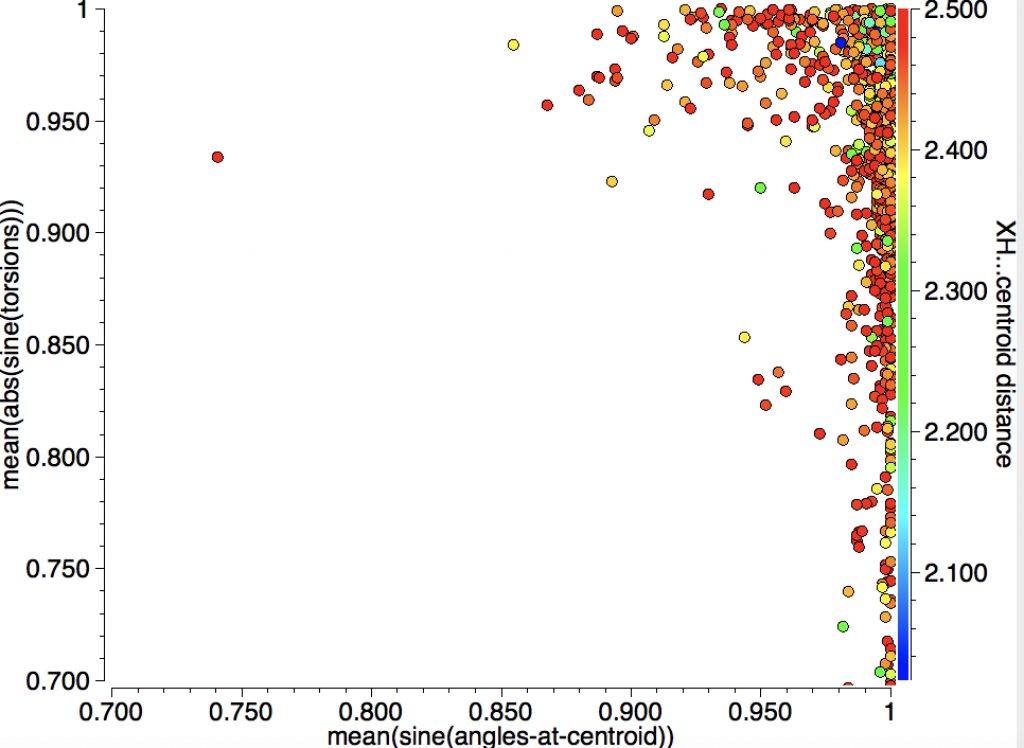
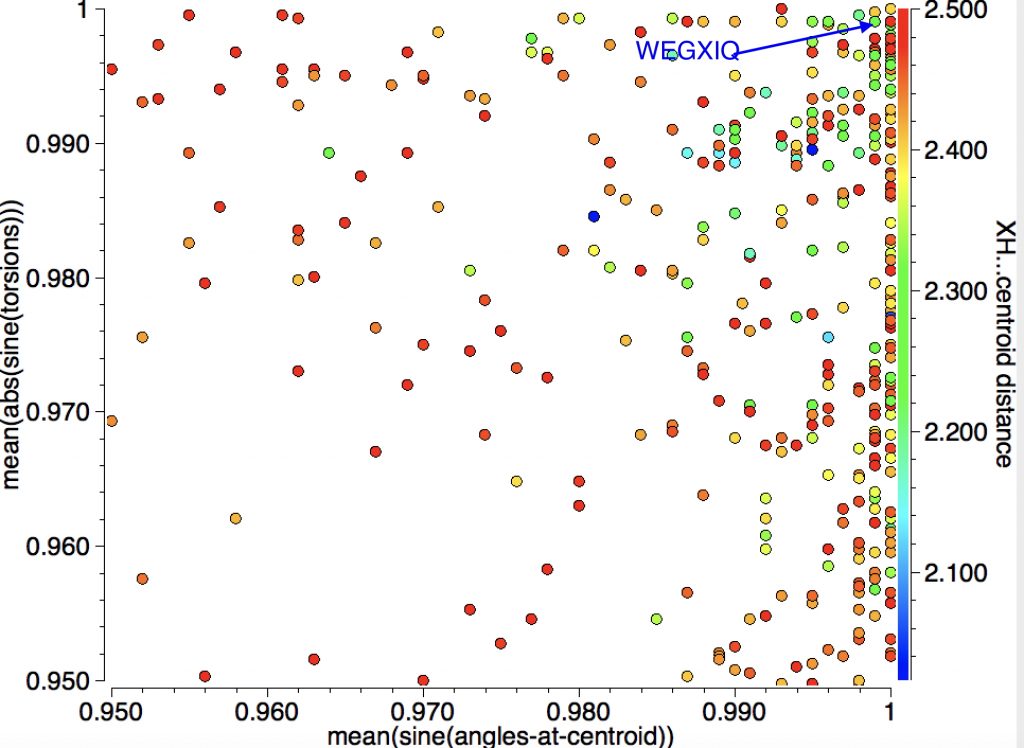
and an expansion:
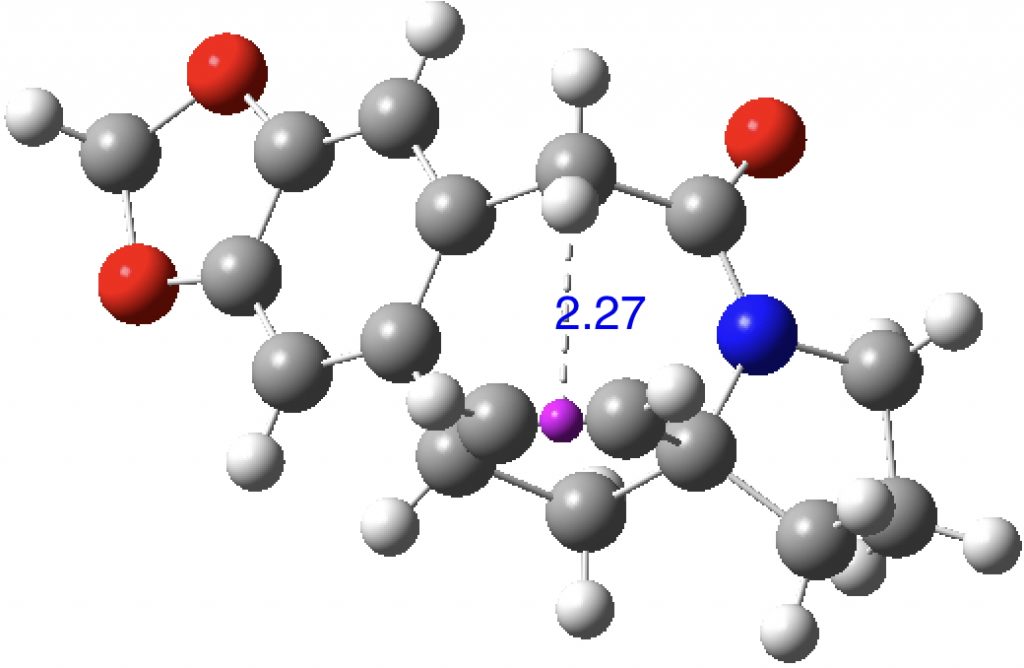
A close CH…π approach is illustrated by WEGXIG (dataDOI: 10.5517/CCNDCBQ):
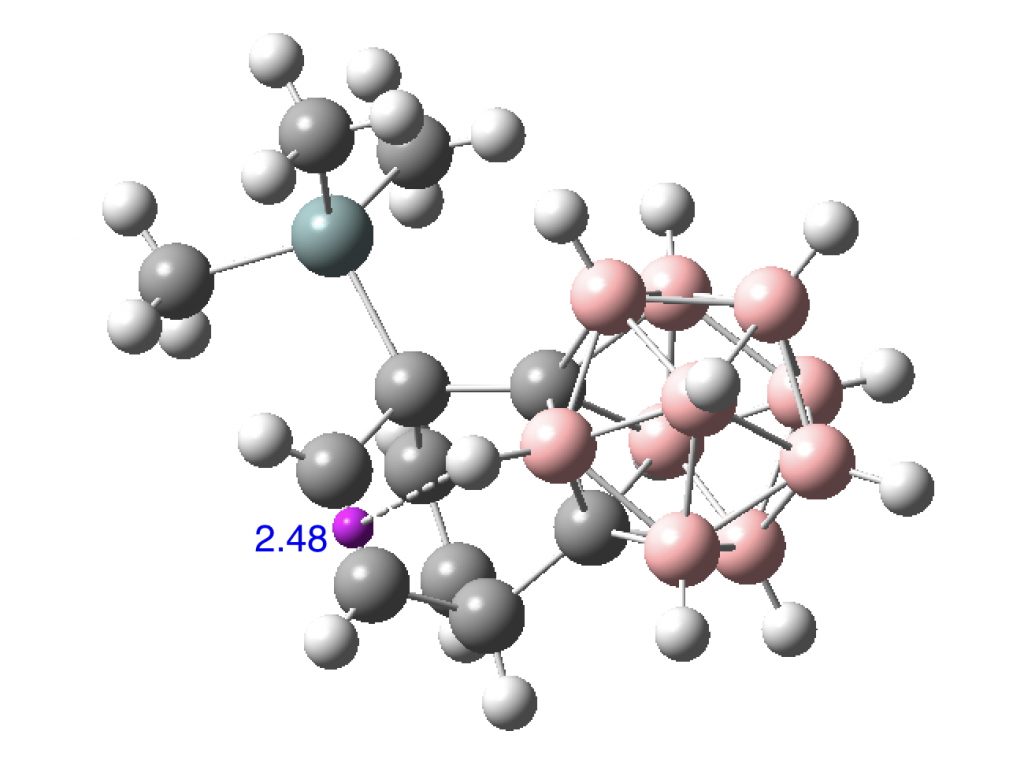
And here is CESZAD for a BH…π approach (dataDOI: 10.5517/CC101BP2 ).
Here is an angle I have hitherto ignored, that subtended at the “acidic” hydrogen. For X-H (X= B,C,N,O.F,Cl,S,P,Si,Br,I,Se) the intermolecular search is shown below followed by the intra-molecular search.
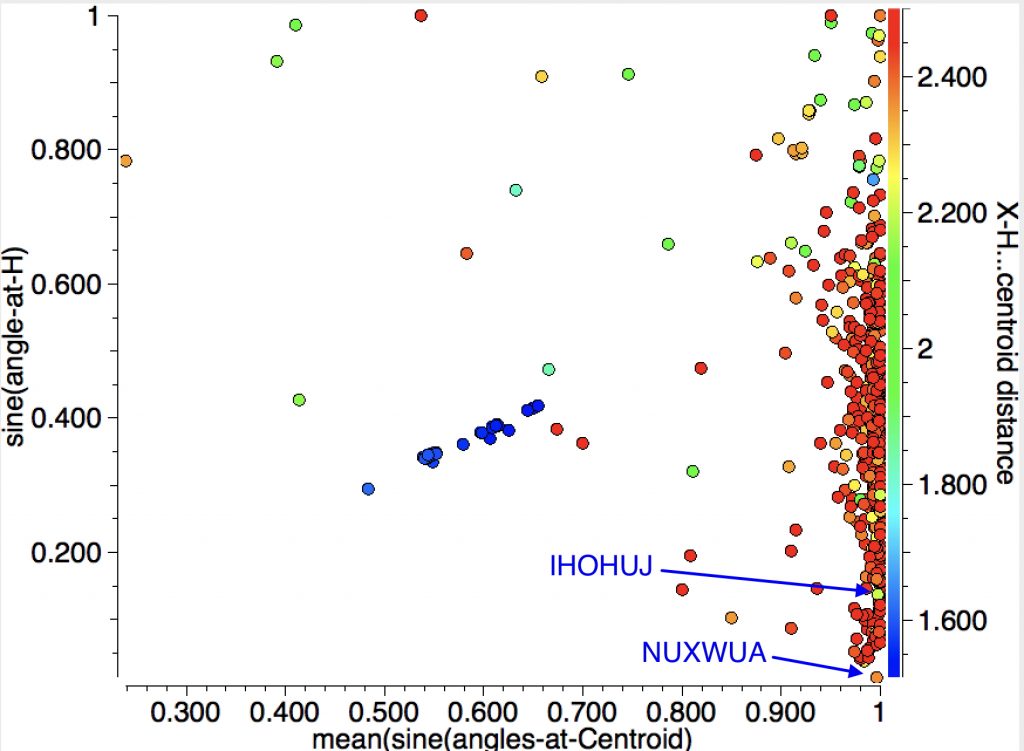
The Y-axis shows the sine of the angle subtended at H; smaller values indicate a more linear X-H-centroid angle.
1. NUXWUA (dataDOI: /10.5517/CC10787R) 2.36Å
2. IHOHUJ (dataDOI: 10.5517/CCQCYLL) 2.21Å
With the intramolecular analysis, we have seen IGELAJ before as having the shortest approach. The X-H-centroid angle is also pretty linear. BONREB (dataDOI:10.1107/S0108270199009919) is almost as short at 2.17Å. Note that the distribution at almost linear angles is sparser for the intra- compared to the inter-molecular cases.