Both the cyclopropenium cation and the cyclopentadienide anion are well-known 4n+2-type aromatic ions, but could the two together form an ion-pair?

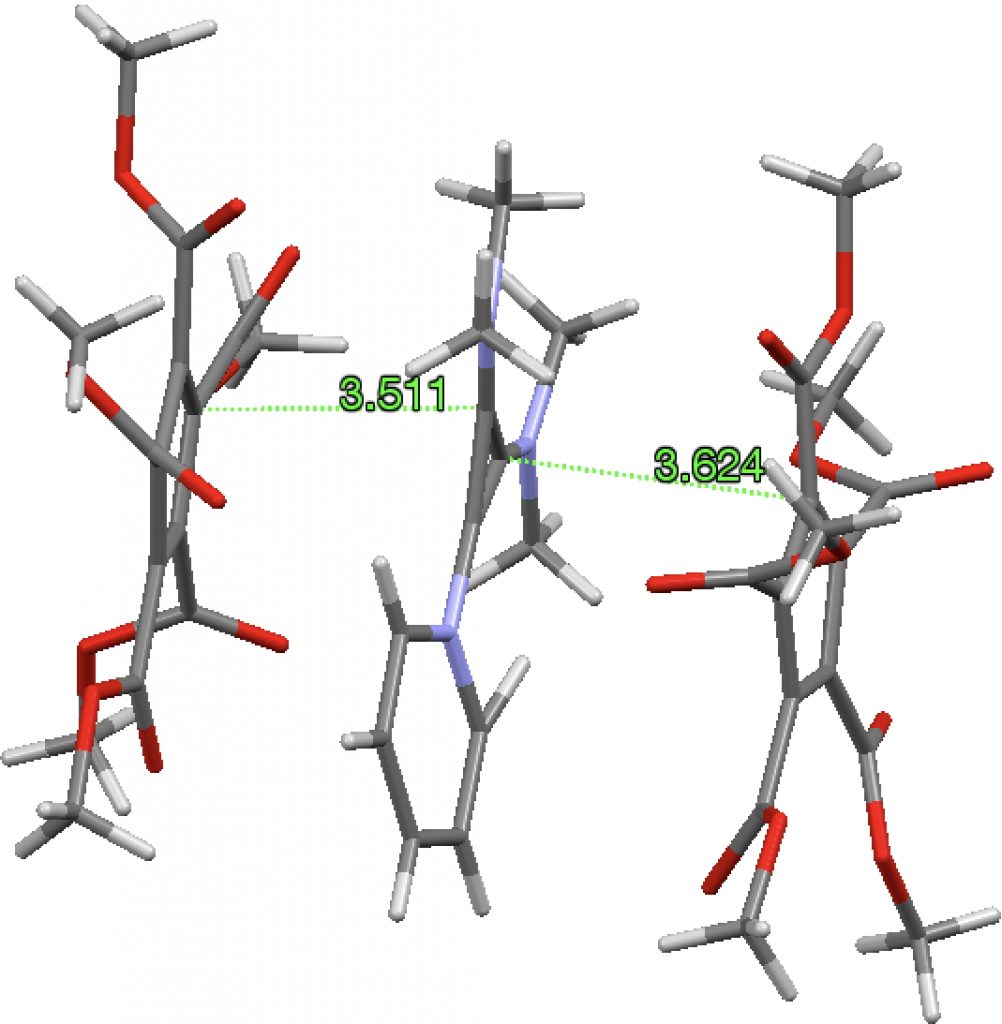
A search of the Cambridge structure database reveals 52 instances of the cyclopropenium cation with a variety of counter-anions, 77 cyclopentadienide anions with a variety of counter-cations and one (SOWMOG, private communication to CSD) where the two sub-structures are common. The pyridinium-cyclopropenium fragment is actually a di-cation stabilized with dimethylamino substituents, with these charges balanced by two cyclopentadienide anions stabilized with ester substituents. The stacking distance between the ion-pairs is ~3.5-3.6Å, a bit larger than normal π-π stacking distances of 3.2-3.3Å
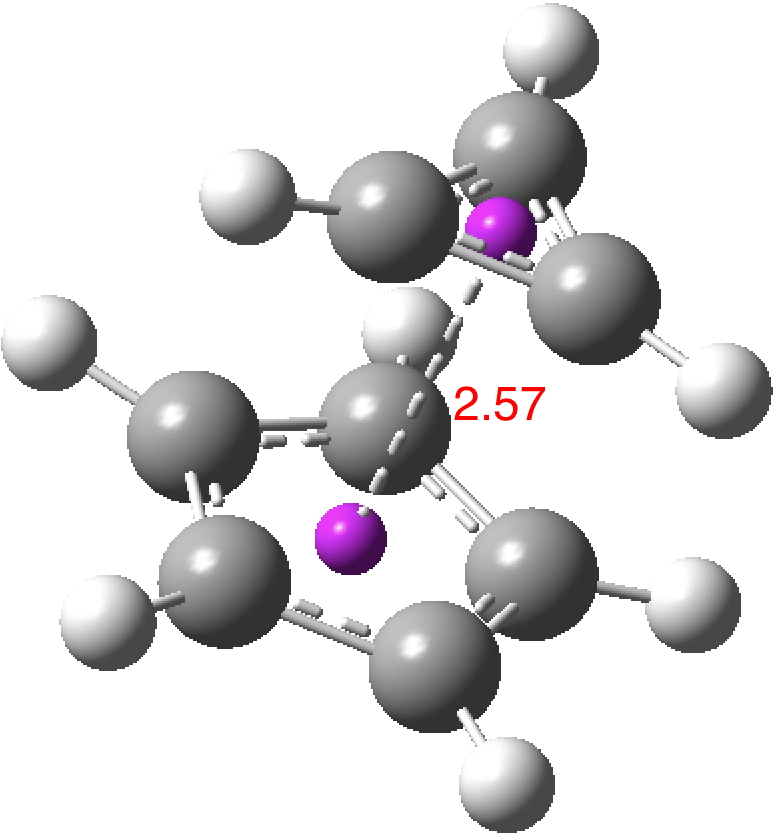
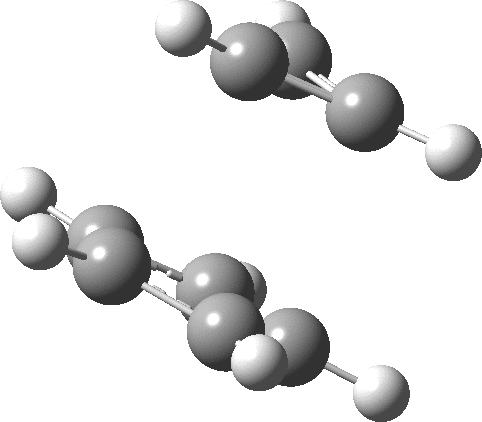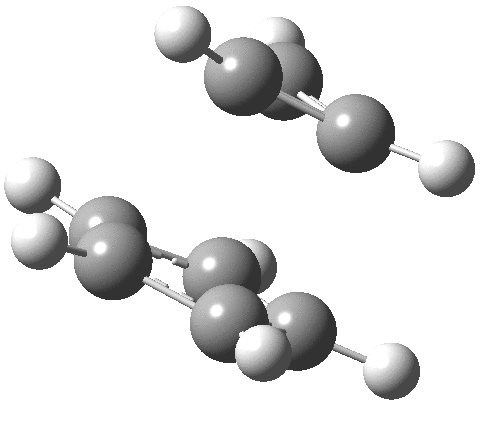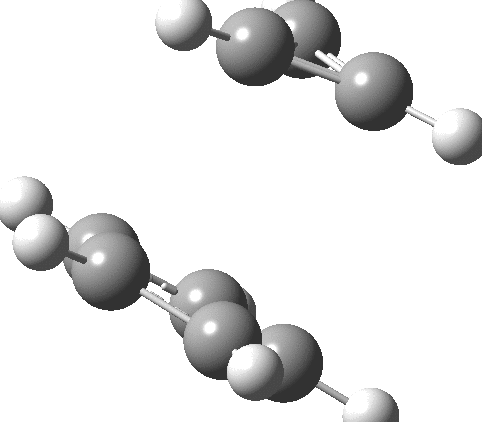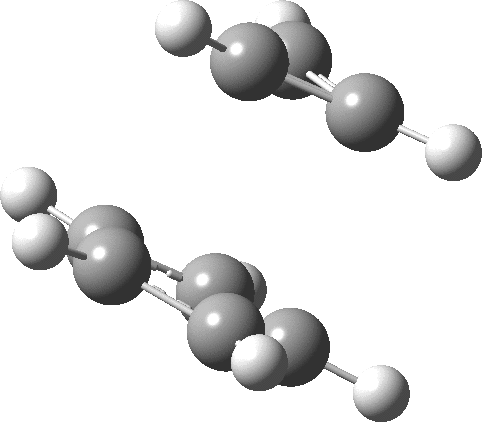
So could a “pure” cyclopropenium cyclopentadienide ion-pair exist, and if so what would its π-π stacking distance be? A ωB97XD/Def2-TZVPPD/SCRF=water calculation (DOI: 10.14469/hpc/2442) provides one answer to this question; 2.57Å!‡ It is a true minimum in the potential energy surface (all +ve force constants) with a calculated dipole moment of only 7.57D. This species is “only” 27.1 kcal/mol higher in ΔG than the neutral hydrocarbon (DOI: 10.14469/hpc/2443), a difference which is as low as it is because of the gain in aromatic stabilization of two rings upon ion-pair formation.
A few posts back, I was considering candidates for the most polar neutral compound synthesized and I suggested a candidate with a dipole moment of ~22D, based as it happens on cyclopropenium and cyclopentadienide rings directly connected by a bond. So when this bond is removed and the two rings are allowed to stack one above the other, we now have an interesting inversion of the original challenge: what is the least-polar ionic organic compound (ionic in the sense of being an unconnected ion-pair)?
Here are some more properties of this intriguing “neutral” ion-pair.
- It has a number of low-frequency modes with correspond to the two rings moving with respect to each other (ν 216 cm-1)
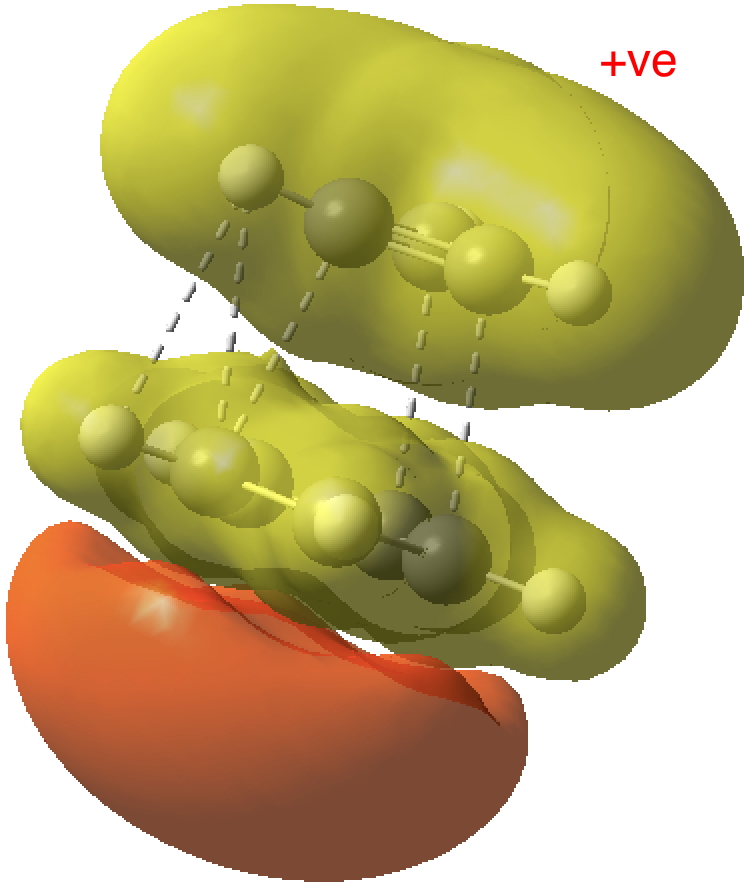
- The molecular electrostatic potential illustrates the sense of polarization, with negative region (orange) residing on the 5-membered ring:
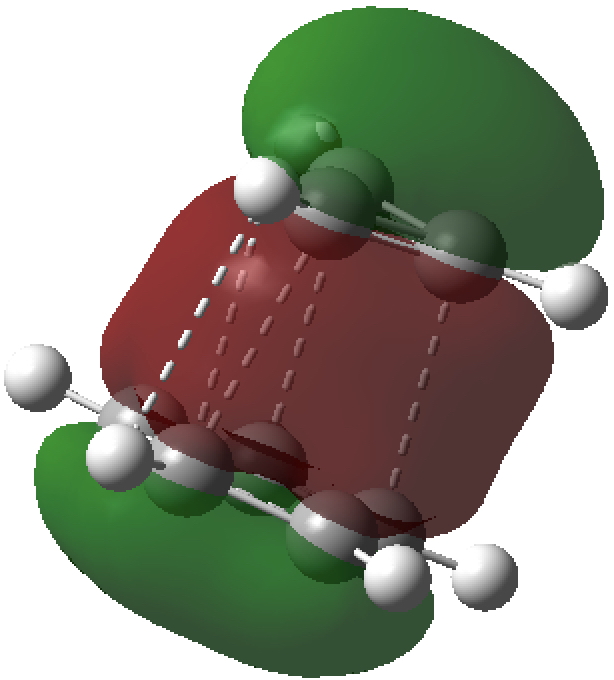
- The most stable π-type molecular orbital (below) reminds of the π-complex formed in the benzidine rearrangement and that in fact modelling this ion-pair may require a multi-reference (CASSCF) wavefunction, with the single-determinantal one used here only being a first approximation.
- A QTAIM analysis of the electron density topology shows only weak “bond” connectors between the two rings, with ρ(r) being typical of weak interactions such as hydrogen bonds.
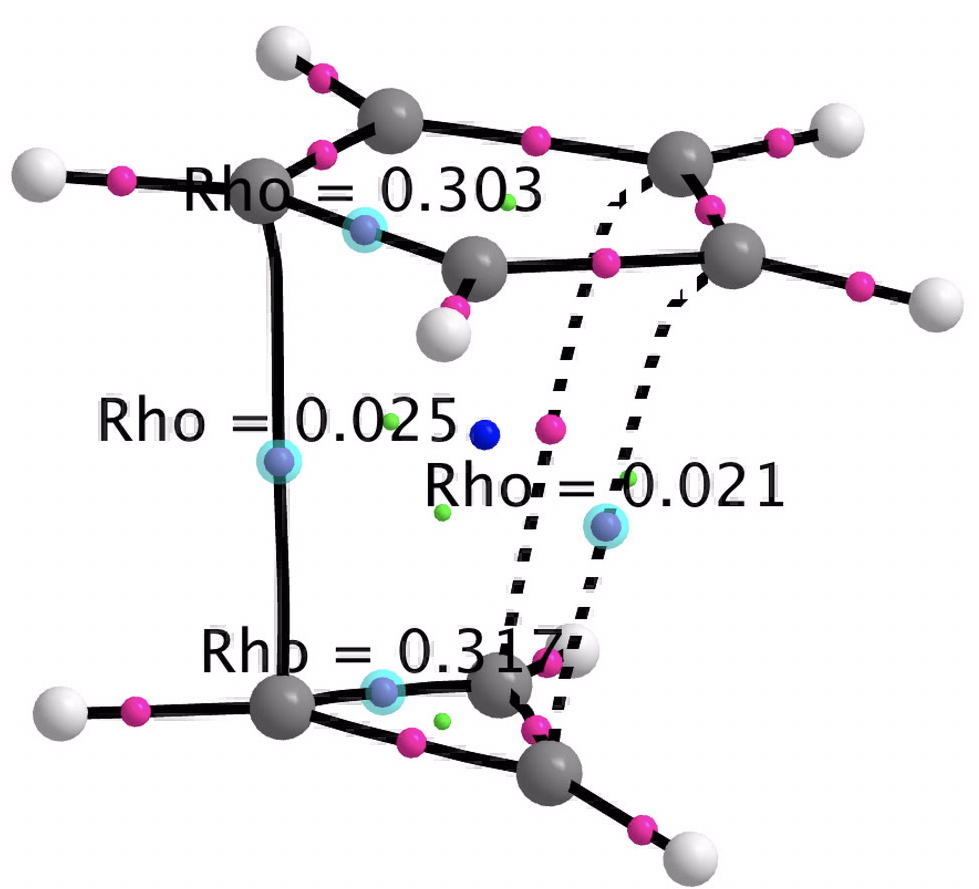
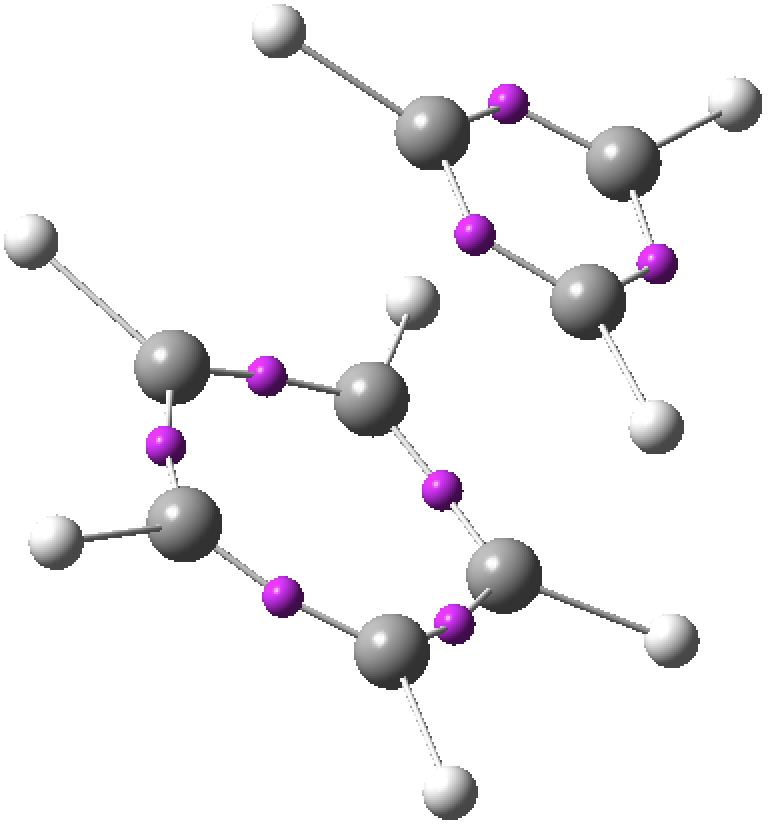
- An ELF (electron localisation function) analysis also holds no surprises, with all the electron density basins (purple) confined to the two rings, just as expected of an ion-pair.
- I will leave one further question to a future discussion; what happens to the aromaticity and ring currents of the two individual rings as they combine to form this ion-pair? Might this property be connected to the very close separation between the two rings?
So we have a remarkably “neutral” ionic hydrocarbon to match the “ionic” neutral organic molecules previously discussed. This ion-pair may yet prove to have interesting properties, even if is unlikely to be synthesized without the addition of stabilising substituents.
‡ For example, the stacking distance in graphite is 3.35Å.
Author
Tags: Anions, Aromatization, Cation–pi interaction, Chemistry, Cyclopentadienyl anion, Ion, Ion association, potential energy surface, Simple aromatic rings
Indeed interesting hypothesis. I computed current intensities for C3H3+, C5H5– and the complex, all in water and at the same level of theory as you had computed.
First of all I am surprised by the large current intensity of C3H3+. I have to check it further to see if a part of that is coming from the σ framework of the molecule. Free C3H3+ has a current intensity of 10.8 nA/T (nano-Ampere per Tesla).
Free C5H5– has a current intensity that is pretty close to that of benzene (13.7 nA/T).
After complexation the current intensity of both systems decrease slightly. In the complex C3H3+ has a current intensity of 9.7 nAT and C5H5– sustains a current of 13.3 nAT.
Thanks Cina.
Are the large diatropic currents associated with the individual rings, or can it be said that they are a “three dimensional” property of the entire complex?
You are welcome. I measured the currents that was passing through the AIM interatomic surfaces just to avoid ambiguity regarding the choice of integration plane. So, these are currents associated with each species. Since they are weakly interacting the current that passes through weak π-π bonds is indeed negligible.
The next homologous ion-pair would be tropylium cyclopentadienide. Two crystalline examples are known; DOI: 10.1002/chem.20160068 (3D model: DOI: 10.5517/CCDC.CSD.CC1K1TYC) and DOI: 10.1071/CH9860165.
The computed properties of the parent system (DOI: 10.14469/hpc/2463, dipole moment 10.6D) show it to be 24.0 kcal/mol higher in ΔG than the covalent non-ionic form (DOI: 10.14469/hpc/2467.
The two rings are rather more separated than the cyclopropenium case, and also translocated.
Here is the UV/Vis spectrum, calculated using ωB97Xd/Def2-TZVPPD/SCRF=water (100 states, DOI: 10.14469/hpc/2451), very pale yellow.
A CASSCF(6,6)/Def2-TZVPP/SCRF=water calculation on the ion-pair optimises to a distance of ~2.8Å (DOI: 10.14469/hpc/2470), but this type of calculation does not account for the dispersion attractions between the rings that might affect the value.