This, the fourth candidate provided by C&EN for a vote for the molecule of the year as discussed here, lays claim to the World’s most polar neutral molecule (system 1 shown below).[1] Here I explore a strategy for extending that record.

The claim for 1 (3 in [1]) is on the basis of its measured dipole moment which is 14.1± 0.7D in THF. This is qualified by the note that the dipole moment might be exalted by complex formation with dimethyl acetamide; the authors report a calculated smaller dipole moment of 9.6D (B3LYP/aug-cc-pVTZ) for the isolated molecule.
Inspection of 1 suggests that it is impossible for both the amino groups to be co-planar with the benzene ring due to steric clashes between the H…H atoms and that they must be twisted to avoid this. If so, the conjugation with the ring would be reduced and so would the charge transfer from the amino groups to the cyano groups (the phenomenon responsible for the polarity). I re-optimised the molecule myself (ωB97XD/Def2-TZVPP/SCRF=THF) and it has C2 symmetry, with both amino groups rotated to avoid those steric H…H clashes (DOI: 10.14469/hpc/1989). The calculated dipole moment (the basis set is a bit better‡ than in [1] and also the geometry is re-optimised in the solvent field) is 13.6D, which is rather closer to the measured value. An alternative explanation for the original mis-match between theory and experiment of 4.5D could be simply the lower quality basis set‡ used in the calculation and no modelled geometric relaxation in the thf solvent field.
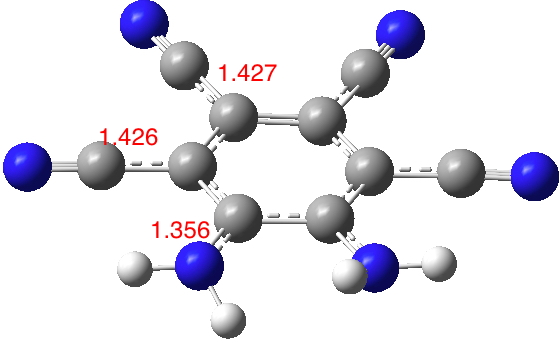
The NC bond lengths shown above will be used as a probe to reveal the extent of conjugation. I tried 2 (R=H), a method of avoiding the steric clash and allowing both amino groups to fully conjugate (DOI: 10.14469/hpc/1987). Note how the amino CN bond length contracts by 0.017Å, whereas the o-cyano CN lengths also contract slightly. The calculated dipole moment for this variation is 16.1D, which seems a rational outcome of increasing the conjugation. However, measured dipole mment values of 10.9 and 12.2D are reported for 2 (5a, R=Me and 5b C7H16) respectively.[1] This is surprising given that these systems avoid any NH…HN steric clash and should therefore allow better conjugation and hence an increased dipole moment. Perhaps it is these molecules rather than 1 where the measured dipole moment is perturbed by other effects?
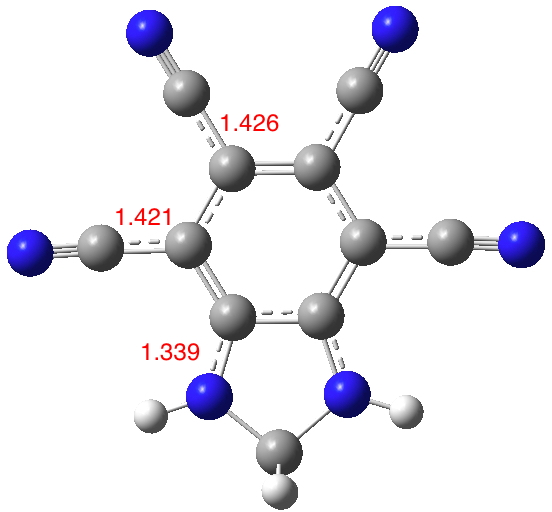
Inspired by these molecules, I thought: why not start with a base aromatic ring that was already polar and sprinkle amino and cyano groups around it? Thus 3 and 4 above. The latter is derived from azulene, which is well-known to have a noticeable dipole moment of its own, with the five ring carrying excess charge to aspire to 6π-electron aromaticity and the seven ring losing charge to again create a 6π-aromatic ring. The cyano and amino groups would serve to stabilize those respective charges.
Firstly 4: Amino groups on the azulene 5,7 positions twist out of plane and do not conjugate (long NC bonds) but all the remaining groups show effective conjugation (DOI: 10.14469/hpc/1988). So one could probably dispense with 5,7-amino substitution. The calculated dipole moment is 21.4D, which elevates the previous value significantly.
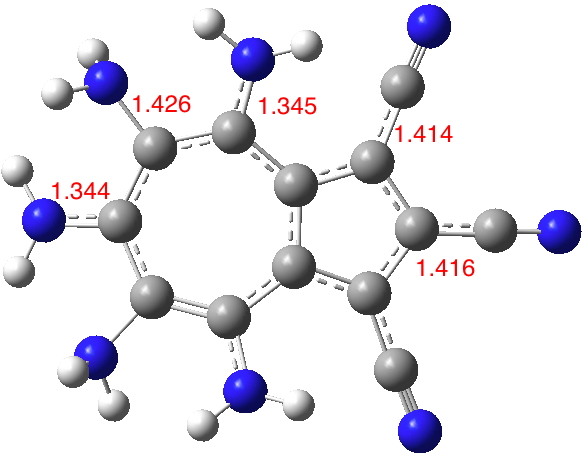
Seven 2- or 3-substituted cyanoazulenes are known in the CSD (Cambridge structure database) and likewise seven 4, 6 or 8 nitrogen substituted derivatives are known. So it should be possible to combine these two groups onto an azulene ring.
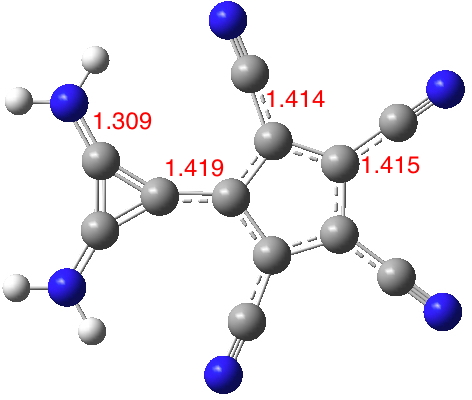
Finally 3, where the amino CN bonds are even shorter, indicating increased stabilization of the cyclopropenium cation ring formed by charge transfer (DOI: 10.14469/hpc/1990). The amino groups no longer clash sterically. The central CC bond (nominally a double bond) is lengthened considerably to facilitate the charge transfer between rings and hence mutual aromatization, the five 5-ring bonds are 1.405Å (typical aromatic values) and the 3-ring 1.367-1.38Å (again aromatic values). This candidate has a dipole moment of 21.7D, despite its smaller size decreasing the separation of the charges and hence the moment.
If the function of a molecule of the year is to inspire ideas in others, this one has certainly achieved its purpose! Now for the syntheses!
‡Anionic systems always benefit from better basis sets, much more so than neutral or cationic molecules.
Author
References
- J. Wudarczyk, G. Papamokos, V. Margaritis, D. Schollmeyer, F. Hinkel, M. Baumgarten, G. Floudas, and K. Müllen, "Hexasubstituted Benzenes with Ultrastrong Dipole Moments", Angewandte Chemie International Edition, vol. 55, pp. 3220-3223, 2016. https://doi.org/10.1002/anie.201508249
Tags: Approval voting, Voting
Below is the calculated TD-DFT (TPSSh) UV/vis spectrum of 1 (DOI: 10.14469/hpc/1992). The optical gap (energy of the first vertical excitation) is ~3.3 eV (experimental value ~3.0 eV). The value reported in the article is 4.0 eV based on the HOMO-LUMO gap of the DFT orbitals.
Here I add the calculated TD-DFT UV/vis spectrum (TPSSh) for compound 3 (DOI: 10.14469/hpc/1991). The first excitation has an energy of 3.85eV, rather higher than 1.
Henry,
Did you calculate dipole moment of N,B-biphenyls?
http://www.ch.imperial.ac.uk/rzepa/blog/?p=1856
Alex:10.7D for the 4,4′ isomer and 8.1D for the 1,1′ B/N form. These are large, but not quite record breaking. Perhaps a 1,1′,4,4′ version might though?
On a rather different point, the original blog noted above contains links (more accurately persistent identifiers or PIDs) to the calculations, dated from 2010. Thus 10042/to-4855 relates to the 4,4′ calculation (this Handle-based PID was also subsequently assigned a more formal DOI form 10.14469/ch/4830 to allow further metadata to be disseminated via DataCite). These were inserted into the post for precisely the reason illustrated by Alex’s question. Thus resolving these PIDs allows access to the original calculation logfile from when the values quoted above come. Because PIDs point to open files, anyone who might be interested can retrieve this data, not just the originator of the calculation.
Dear Prof. Rzepa,
Please note that the methodology of the experimental measurements on the molecules of ref 1 is based on the fact that it tries to remove the solvent effects and provide the permanent dipole moment. (See ref 21, 22, of your ref. 1.) Thus the calculated dipole moments should be done in vacuo. Calculated dipole moment for one molecule is also present in ref. 1. (see page 3222, column 2, paragraph 1)
I think that the experimental dipole moment is high because of the DMAC. By adding the THF in implicit solvation you come closer to the experimental value. But this is done by the addition to the solute of two different phenomena. In our experiment because of the DMAC and by your calculation because of the implicit solvation (THF).
The basis sets employed in ref 1 and in your calculation accounts for such a big difference?
I think not. Dipole moment is a ground state property and it should not give such a big difference.
Georgios,
Thanks for your feedback.
I am not convinced that the a dipole moment measured in thf should be treated as if in vacuo using theory. The solvent field will affect the calculated geometry, and this perturbation to the geometry cannot be ignored. For dipole moments >10D, this geometric perturbation can be quite significant.
Anionic systems are also recognised as requiring special diffuse basis functions added. I agree that compactifying the electron density by including instead a solvent field is another solution to the problem. But I think to properly model charge separation into an anionic region it is useful to use a high quality basis set AND a solvent field. Nowadays there is no computational reason for not extending a double-ζ basis (aug-cc-pvdz) into triple-ζ with good polarization functions.
Overall, I still think that pinning back the two amino groups with a carbon bridge should result in an increase and not a decrease in the conjugation and hence in the charge separation, as measured with a dipole moment. So this aspect does need some explanation.
At any rate, I was really inspired by your work to suggest that a dipole moment of ~14D might be increased by further molecular design. A full-blown ion pair (a “neutral” system) can sustain dipole moments between 20-35D, and that may be the ultimate ceiling.
Dear Prof. Rzepa,
I don’t disagree that the solvent will affect the geometry of the solute.
But the theoretical framework of the experimental treatment (Ref. 21, 22) removes the effects of the solvent on the solute and this is what it is reported in the paper. Thus the comparison must be made with the permanent dipole moment of the molecule and not with the sum of the permanent and the induced by the solvent dipole moment. If we follow your suggestion then the difference between experimental and theoretical values for the other molecules (that do not have DMAC attached) will be 4 D. Try to do these calculations and I hope that the results will convince you. If not come back to me and we will discuss it further.
Regarding the different basis sets: The basis set you suggest cannot justify such a big difference. Try it your self. The difference on the molecules of the paper in vacuo employing the same functional must be 0-0.2 D.
Thanks for your comments.
I hope your inspiration to be “synthesizable””!!!
All the best,
Georgios
Here is an article which I think also presents nicely some of the challenges of studying dipole moments as both “isolated” entities and in “bulk” (DOI: 10.1007/430_2012_78).
Table 4 in particular suggests that the dipole moment of an isolated amino acid is ~11.1D and when the “bulk” is simulated by an 8Å cluster of point charges and dipoles (similar to the continuum solvent fields) it increases to ~16.3D (alanine). These values are remarkably close to the values I reported above.
If one is consistent in eg quoting say “bulk” values, then presumably one can compare structural trends, such as the difference between a 1,2-diamino benzene (example 1 above) and a variation which is free of steric H…H clashes (2 above).
So the crucial point needing a conciliation between theory and experiment is the following.
Theory suggests that when the 1,2-diamino system is “pinned back” to remove the H…H di-amino steric clash, allowing both amino nitrogen lone pairs to be coplanar (π-π-overlapping) with the benzene ring, both the conjugation and the resulting dipole increase. One would expect this to manifest for the isolated molecule as well as under bulk conditions.
Experiment seems to suggest that this structural change has little or no effect on the dipole moment.
The issue then is whether these two observations can be reconciled?
I did not manage to download the paper you suggest.
But:
Have you considered the “packing” factor in crystals?
π-π interactions?
Just an idea.
Georgios,
The article above has the title “Reliable Measurements of Dipole Moments from Single-Crystal Diffraction Data and Assessment of an In-Crystal Enhancement” (I have checked the DOI and it is correct), and perhaps I can quote part of the abstract here: The question whether a molecular dipole-moment enhancement in the solid state is fact or artifact is studied by a number of techniques: A theoretical molecule embedded in a cluster of point-charges gives a substantial enhancement, in agreement with Hirshfeld atom refinement with point charges and dipoles. The experimental techniques, multipole refinement and wavefunction fitting, lead to smaller dipole-moment enhancements than the theoretical predictions.
It does appear that various models for obtaining dipole moments and in particular enhancements in the “bulk” can indeed disagree, and so there must be a big question mark about whether “bulk” enhancement is real or an artefact.
This article actually studied zwitterionic amino acids, where π-π stacking is not normally an issue, but where “point charges” are probably the dominant effect.
Of course one may also discuss whether an internally-ionized amino acid is truly a “neutral molecule” and whether internal dipole brought about purely by electron redistribution is equivalent to full ionization brought about by proton tautomerism. Perhaps the neutral molecules discussed in this post and the ionic amino acids (an example is computed in the following post) are just two points in a continuum of polarity.
I do have some questions about the cyanoaminobenzenes. How much higher in energy are the tautomers in which a hydrogen moves from the amine to the cyano group? I presume this would result in extreme loss of aromaticity? One could also calculate that aromaticity with the amino groups co-planar and orthogonal and compare the results? The examples I suggested do the precise opposite, ie the movement of charge enhances the aromaticity (of two rings) rather than decreases it as the driving force; it is polarity driven by aromatisation.
Henry,
There is a simple way to improve dipole moment of a molecule in computing experiments: to increase distance between “+” and “-” parts. For example, for 4-B,4′-N-biphenyl BC5H5-(C6H4)n-C5NH5
n=0 10.8 D
n=1 16.8 D
n=2 22.8 D
n=3 28.8 D
Alex,
Is that a simple coulomb law calculation or full SCF values?
I should imagine that as the band gap (HOMO-LUMO energy difference) narrows with increasing separation; so the closed shell wavefunction must eventually become unstable towards lower energy open shell states which would have of course much lower polarity/dipole moments.
The Cambridge structure database does not seem to have any examples of bis-aryls with one B in the 4′ position. The only examples are 1,1′ regioisomers.
Henry,
It is SCF values for optimized geometries.
Yes, HOMO-LUMO gap is narrow and separation is decreased. And nonpolar H2B—N tautomer becomes more stable then HB—NH (in gas phase).
n HOMO-LUMO E(H2B—N)-E(HB—NH)
0 1.7 eV 19 kcal/mol
1 0.9 eV 6 kcal/mol
2 0.5 eV -2 kcal/mol
3 0.2 ev -7 kcal/mol
_
Thanks Alex. I suspect that n=1-3 are all probably biradicals then.