The journal of chemical education has many little gems providing inspiration for laboratory experiments. Jonathan Piard reports one based on the reaction below[1]; here I investigate the mechanism of this transformation.

There are two things going on here; an electrocyclic ring opening involving breaking the C-O bond, with a cis/trans isomerism of the alkene (concurrent or consecutive). A crystal structure of the dinitro analogue establishes the trans stereochemistry[2]. This product zwitterion is highly coloured (blue-purple) unlike the colourless reactant. The rate at which this colour clears can be easily measured in a UV/visible spectrometer, and from this activation parameters are inferred as a function of the solvent.
Photochemically, this reaction is too complex to study quickly using computation, but the thermal back reaction is much easier. Applying the ωB97XD/6-311G(d,p)/SCRF=solvent procedure and using the C-O bond as a reaction coordinate results in the following transition state[3] IRC profile[4] for DMSO as solvent, here connecting to the cis-alkene. The thermal forward barrier for the C…O cleaving is ~12 kcal/mol. More significantly, the back reaction is only <2 kcal mol, which is very much lower than that reported[1] (~22 kcal/mol) and makes the cis-alkene very much a transient species and therefore not the coloured species being measured.

A second transition state involving C=C bond rotation is located[5] and this yields the following IRC[6] to form the trans-alkene, with activation parameters listed below. It is entirely probable however that the forward photochemical reaction follows a different course involving conical intersections; an interesting study in its own right, but beyond the scope of this post.
| Measured and computed activation parameters for the thermal back reaction | |||
|---|---|---|---|
| Solvent | ΔG298, kcal mol-1 | ΔH, kcal mol-1 | ΔS, cal K-1 mol-1 |
| DMSO, measured | 22.0 | 26.4 | +14.6 |
| toluene, measured | 19.0 | 14.3 | -15.6 |
| DMSO, calc[5] | 21.2 | 20.4 | +2.6 |
| toluene, calc[7] | 18.6 | 19.1 | +1.8 |
The next issue surrounds the effect of solvent. Most spectacular are those of the activation parameters for the thermal back-reaction. The measured activation entropy ΔS changes with solvent by Δ30.2 cal, and the enthalpy ΔH by Δ12.1 kcal, which are enormous solvent effects. Are they in fact real? It is reassuring at least that the calculated free energy agrees pretty closely with the measured values. So too does the decrease in ΔG in changing the solvent from DMSO to toluene (3.0 kcal/mol measured, 2.6 calculated).
There is little sign of a large solvent effect in the calculated values. This could be for three reasons.
- The first is that adding explicit, hydrogen-bonded solvent molecules to the system is essential. As the zwitterionic character is lost when the trans-alkene starts to rotate, the system will become less ionic, and hence shed solvent molecules and gain entropy. If the solvent is not capable of hydrogen bonding, as say toluene, these will not be shed and the entropy will not increase at the transition state. It is difficult however to reconcile this picture with the apparent large loss of entropy for toluene as solvent.
- The second reason is because of a complete change in mechanism, one not modelled here.
- For completeness, one should also mention that the measured values might simply be in error, either due to typographical mistakes or indeed in numerical analysis.
I will conclude with the colour. It is possible to compute the electronic excitations across the range 180-580nm, and I show below a difference spectrum (for DMSO as solvent) with the product +ve and the reactant -ve. You can see the spectacular red-shift for the highly conjugated zwitterion! The absolute value of λmax is not red-shifted enough (by about 65 nm), but the effect remains real.
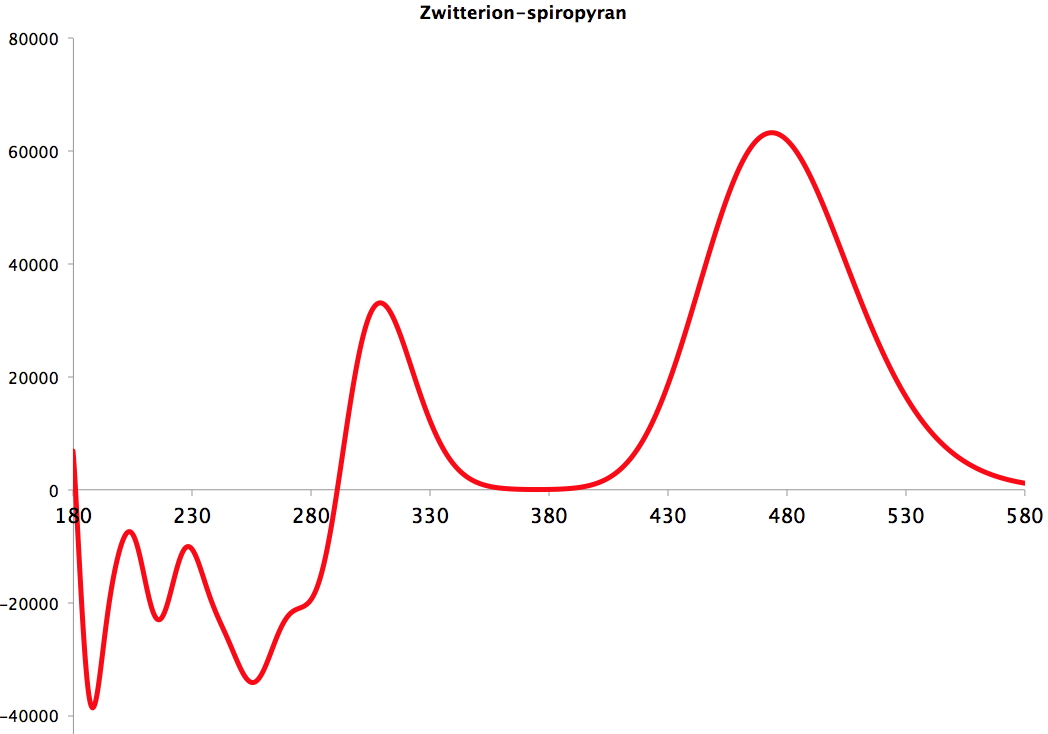
The above experiment is for undergraduate chemistry laboratories. I suggest that a computational reality check could also easily be included into such a lab, and would certainly help give students a broader perspective.
Author
References
- J. Piard, "Influence of the Solvent on the Thermal Back Reaction of One Spiropyran", Journal of Chemical Education, vol. 91, pp. 2105-2111, 2014. https://doi.org/10.1021/ed4005003
- J. Hobley, V. Malatesta, R. Millini, L. Montanari, and W. O Neil Parker, Jr, "Proton exchange and isomerisation reactions of photochromic and reverse photochromic spiro-pyrans and their merocyanine forms", Physical Chemistry Chemical Physics, vol. 1, pp. 3259-3267, 1999. https://doi.org/10.1039/a902379h
- H.S. Rzepa, "C 19 H 18 N 2 O 3", 2015. https://doi.org/10.14469/ch/189540
- H.S. Rzepa, "C19H18N2O3", 2015. https://doi.org/10.14469/ch/189573
- H.S. Rzepa, "C 19 H 18 N 2 O 3", 2015. https://doi.org/10.14469/ch/189546
- H.S. Rzepa, "C19H18N2O3", 2015. https://doi.org/10.14469/ch/189648
- H.S. Rzepa, "C 19 H 18 N 2 O 3", 2015. https://doi.org/10.14469/ch/189579
Tags: calculated free energy, chemical education, Jonathan Piard