One thing leads to another. Thus in the previous post, I described a thermal pericyclic reaction that appears to exhibit two transition states resulting in two different stereochemical outcomes. I noted that another such reaction appeared to be a [1,6] carousel migration in homotropylium cation,[1] where transition states for both retention and inversion of the configuration of the migrating group (respectively formally allowed and forbidden) were reported (scheme below). Here I explore this system further.  Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array.
Firstly, the pathway leading to inversion.[2] The reaction path (ωB97XD/6-311G(d,p)/SCRF=chloroform) has got a very odd (table-top mountain) shape, whereby the region of the transition state (IRC = 0.0) is very flat, and the region close to reactant and (identical) product is very steep. The gradient norm shows this best, with sharp spikes at IRC ± 4.2. Something clearly is happening here to cause this behaviour. Before moving on to analyze this, I want you first to observe the methyl groups below. Note how one of them rotates at the start of the process, and the other at the end. I have elsewhere called this behaviour the methyl flag, and it is due to stereoelectronic re-alignments of the C-H groups accompanying the changes in the conjugated array. 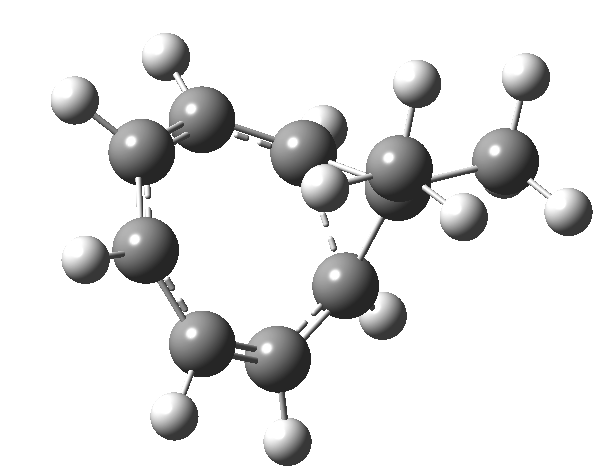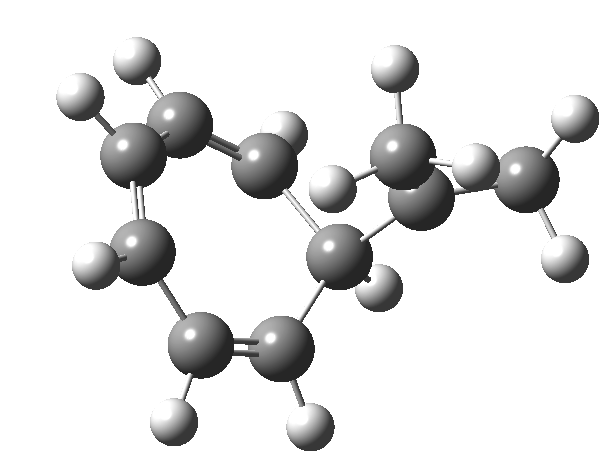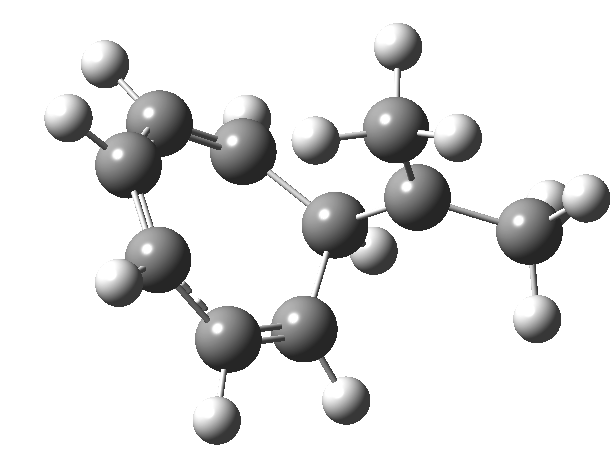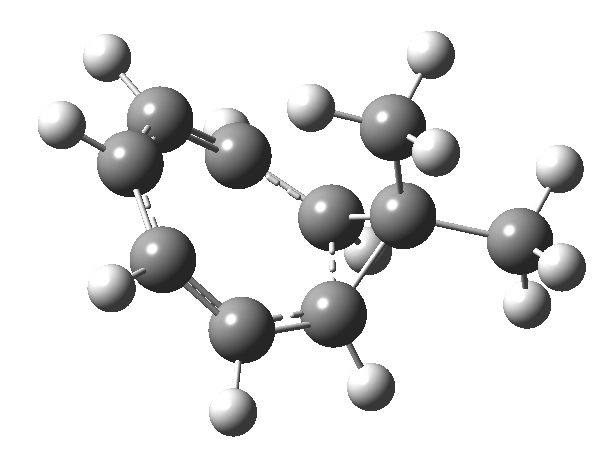

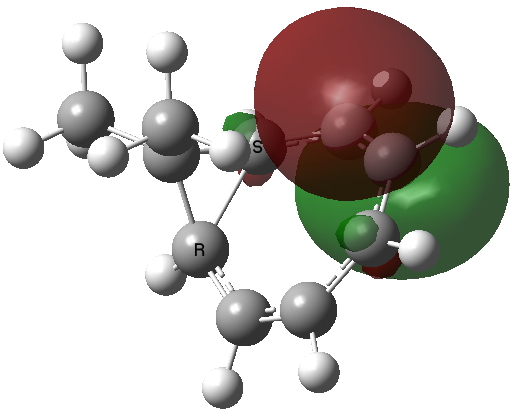
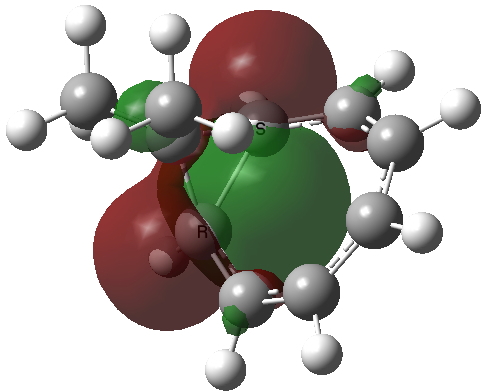
 The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
The homotropylium cation is said to be homoaromatic, indicating that cyclic conjugation can be maintained across a ring in which the σ framework is interrupted at one point. A NICS probe placed at the ring critical point of this molecule reveals a chemical shift of -11.3 ppm[3], very similar to eg that obtained for benzene itself. The three highest doubly occupied NBOs (below) show two normal π-type orbitals and one rather different one that spans the homo-bond (the MOs, before you ask, are a bit of a mess, with lots of mixed contributions from other parts of the σ framework).
| HONBO (two) | HONBO-2 |
 Click for 3D |
 Click for 3D |
At the transition state for the [1,6] migration, the same NICS probe registers a value of +2.6 ppm[4], now firmly in the non-aromatic zone. So this reaction is characterised by two zones, ring-aromatic ones at the start and the end of the process, and a higher energy non-aromatic one in the middle of the reaction pathway as ~enclosed by the region of IRC ± 4.2. The homo-bond in the aromatic zone starts with a length of 1.74Å, reduces to 1.53Å at the transition state and ends up as a normal aromatic bond of length 1.41Å. Meanwhile, the relocated homo-bond changes in the opposite sense, starting as a normal aromatic length of 1.41Å, becoming 1.53Å at the transition state and ending as a homo-length of 1.74Å. Presumably, virtually full strength homoaromaticity can be sustained for a homo-bond of 1.74Å, but as that bond mutates to a σ-bond of 1.53Å, the cyclic conjugation falls off the edge of the cliff, to be restored only at the end. Pericyclic reactions are themselves said to sustain aromatic transition states,[5] and so a simplistic way of looking at this is that the “aromaticity” relocates (or morphs) from the reactant to the transition state, and then back again during the course of the migration. A reaction path from which one can indeed learn a lot.
Now to the pathway in which the migrating group retains configuration. This is no longer a single step concerted reaction,[6] since at the half-way point we no longer have a transition state but a shallow intermediate (~IRC +2, [7]). It (formally at least) becomes a two-step non-concerted process, and the overall barrier is ~5 kcal/mol lower than for the inversion path. The aromaticity changes in a similar manner to before (i.e. IRC ~-5).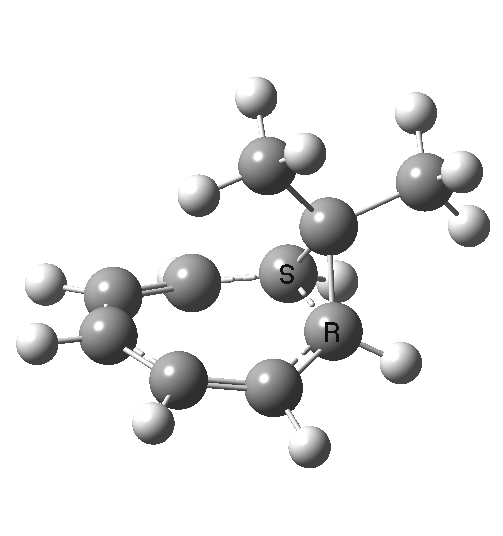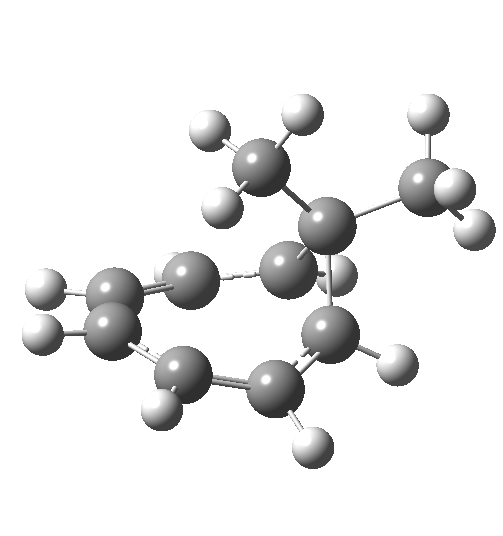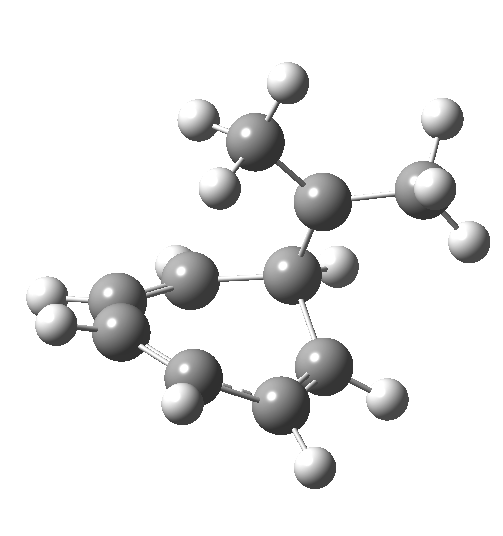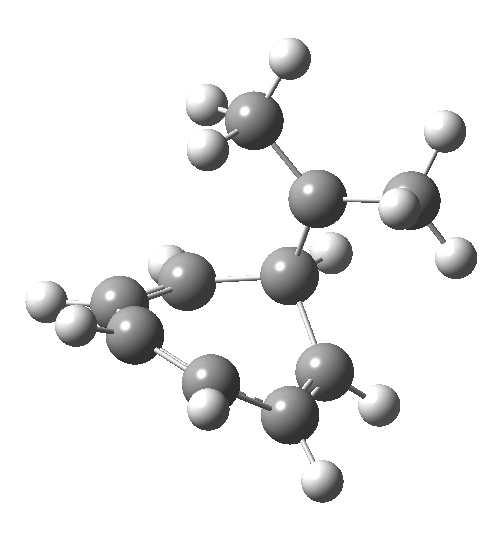


So this emerges as not quite the example I thought it was, but nonetheless unusual with the “forbidden” pathway being concerted and the “allowed” pathway being non-concerted. Molecular dynamics on these two systems would indeed be interesting to see what proportion of the trajectories go via each pathway.
Author
References
- A.M. Genaev, G.E. Sal’nikov, and V.G. Shubin, "Energy barriers to carousel rearrangements of carbocations: Quantum-chemical calculations vs. experiment", Russian Journal of Organic Chemistry, vol. 43, pp. 1134-1138, 2007. https://doi.org/10.1134/s1070428007080076
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134556
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135694
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135695
- H.S. Rzepa, "The Aromaticity of Pericyclic Reaction Transition States", Journal of Chemical Education, vol. 84, pp. 1535, 2007. https://doi.org/10.1021/ed084p1535
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1135668
- H.S. Rzepa, "Gaussian Job Archive for C10H13(1+)", 2014. https://doi.org/10.6084/m9.figshare.1134559
Tags: chemical shift, higher energy, Sangean Table Top Portable Audio Device
Great!
Formerly I found other TS for the pathway leading to inversion (with more high barrier)
Great!
Formerly I found other TS for the pathway leading to inversion (with more high barrier)
Alex sent me his results a few years back, and I was fascinated, since locating both the allowed and forbidden TS allowed some energetic estimate of how “forbidden” the second pathway actually was. It is of course forbidden only in terms of energy. Mind you, in a strict single reference method, where only one closed shell configuration is used, the forbidden pathway often corresponds to an avoided crossing of two configurations/states, which is not modelled.
We therefore do have to remember that “forbidden” pathways are a concept which does not match well with a single configuration closed shell wave function.
Anyway, the location of two “forbidden” transition states is even more interesting. Are they both true pericyclic reactions? As a “forbidden” reaction, is the transition state anti aromatic? How much so? Are both transition states STABLE to open shell configurations?
I do not think this topic is quite exhausted yet.
I note that for my TS, both forming and cleaving bonds in the sigmatropic reaction are ~2.5Å. For Alex’s TS, they are 1.7Å.
I believe similar observations have been made for the [3,3] sigmatropic Cope rearrangement. The “short bond” TS can be considered bis-homoaromatic, and indeed by suitable substitution, such a homoaromatic TS can be stabilised out to become an actual intermediate; see http://www.ch.imperial.ac.uk/rzepa/blog/?p=7756. The analogy however does not work with the current [1,6] sigmatropic.
I have obtained the IRC (previously obtained by Alex), at the ωB97XD/6-311G(d,p)/SCRF=chloroform level.
Whereas the lower energy path was almost flat in the TS region, this one is the inverse, being very sharp! The DOI for this IRC is 10.6084/m9.figshare.1141695.