Ribulose-1,5-bisphosphate reacts with carbon dioxide to produce 3-keto-2-carboxyarabinitol 1,5-bisphosphate as the first step in the biochemical process of carbon fixation. It needs an enzyme to do this (Ribulose-1,5-bisphosphate carboxylase/oxygenase, or RuBisCO) and lots of ATP (adenosine triphosphate, produced by photosynthesis). Here I ask what the nature of the uncatalysed transition state is, and hence the task that might be facing the catalyst in reducing the activation barrier to that of a facile thermal reaction. I present my process in the order it was done‡.
 Firstly, I will hypothesize that since C3 needs to lose a hydrogen, the easiest way of doing so is to form the enol of Ribulose-1,5-bisphosphate. I am going to start by reducing the above model to its core; C1 and the attached phosphate is replaced by a methyl, and C4-5 likewise. In this model, it takes 13.1 kcal/mol of free energy to enolize.[1],[2] This species can then react with CO2 (potentially with an accompanying proton transfer) to give 3-keto-2-carboxyarabinitol 1,5-bisphosphate directly. The transition state at the ωB97XD/6-311G(d,p)/SCRF=water level[3] has an IRC (intrinsic reaction coordinate)[4] that reveals the activation barrier is ~17 kcal/mol with respect to the enol (19.5 in ΔG298), with the overall reaction[5] being exo-energic by -2.6 kcal/mol with respect to the enol, but endo-energic by +10.5 kcal/mol with respect to keto-Ribulose-1,5-bisphosphate + carbon dioxide. Note the characteristic feature at IRC -3.0 of a hidden zwitterionic intermediate, which marks a belated proton transfer occurring AFTER the transition state for C-C bond formation. The reaction is asynchronous for this basic model.
Firstly, I will hypothesize that since C3 needs to lose a hydrogen, the easiest way of doing so is to form the enol of Ribulose-1,5-bisphosphate. I am going to start by reducing the above model to its core; C1 and the attached phosphate is replaced by a methyl, and C4-5 likewise. In this model, it takes 13.1 kcal/mol of free energy to enolize.[1],[2] This species can then react with CO2 (potentially with an accompanying proton transfer) to give 3-keto-2-carboxyarabinitol 1,5-bisphosphate directly. The transition state at the ωB97XD/6-311G(d,p)/SCRF=water level[3] has an IRC (intrinsic reaction coordinate)[4] that reveals the activation barrier is ~17 kcal/mol with respect to the enol (19.5 in ΔG298), with the overall reaction[5] being exo-energic by -2.6 kcal/mol with respect to the enol, but endo-energic by +10.5 kcal/mol with respect to keto-Ribulose-1,5-bisphosphate + carbon dioxide. Note the characteristic feature at IRC -3.0 of a hidden zwitterionic intermediate, which marks a belated proton transfer occurring AFTER the transition state for C-C bond formation. The reaction is asynchronous for this basic model.
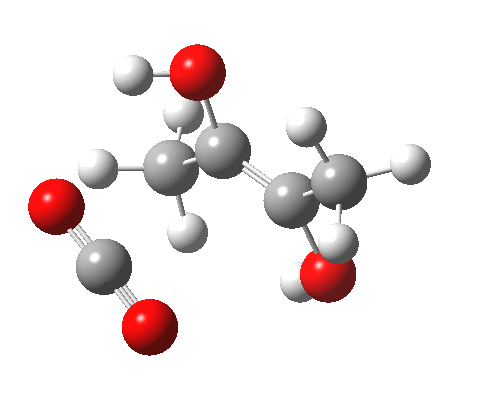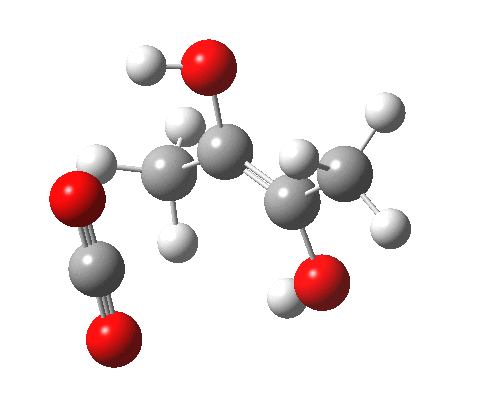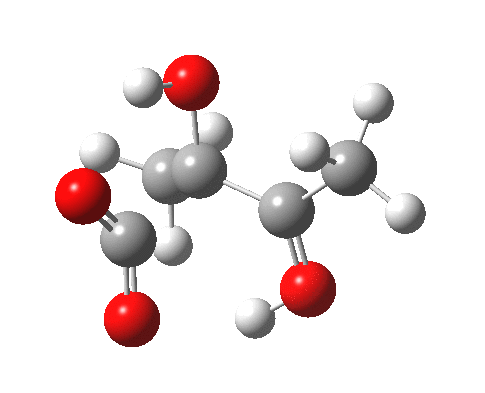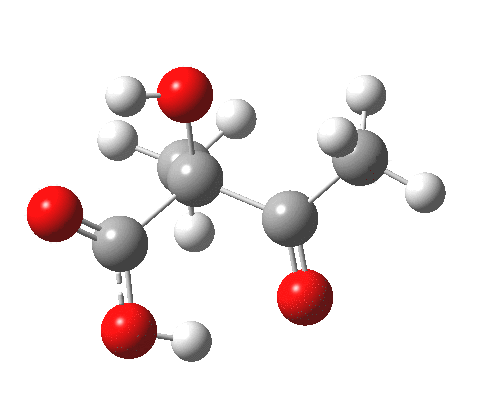


For this very basic (phosphate-free) model of Ribulose-1,5-bisphosphate, the total computed free energy barrier@298K is 32.6 kcal/mol (standard state of 0.041M; reduced by ~1.9 kcal/mol for more concentrated, e.g. 1M solutions). This is ~13 kcal/mol too high to correspond to a uncatalysed fast process at room temperatures, a gap that the phosphate end-groups and the enzyme have to address (a challenge typically enzymes do manage to achieve).
With a basic model in place, it is time to restore those truncated phosphate end-groups to see what their contribution might be (treated as dianions each for the time being, and stabilized by using a continuum solvent field for water). First, the energies:
| System | ΔΔG | Data DOI |
|---|---|---|
| Ribulose-1,5-bisphosphate as keto + CO2 | 0.0 | [6] |
| Ribulose-1,5-bisphosphate as enol + CO2 | 13.0 | [7] |
| Transition state | 34.8 | [8] |
| Acyclic 3-keto-2-carboxyarabinitol 1,5-bisphosphate | 11.5 | [9] |
| Cyclic 3-keto-2-carboxyarabinitol 1,5-bisphosphate | -7.3 | [10] |
Note the network of hydrogen bonds formed at the transition state geometry (below) and the various gauche stereo-electronic alignments[11] which you should really explore in the Jmol 3D model invoked by clicking below.
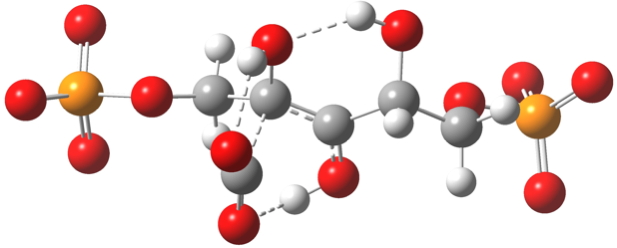
Click for 3D
- Addition of the phosphate groups has little effect on the energetics of the keto/enol equilibrium,
- or on the barrier to reaction with carbon dioxide.
- But, they DO provide a new low energy sink I have not seen described before for the reaction (below), which makes the overall process from Ribulose-1,5-bisphosphate + CO2 exo-energic by -7.3 kcal/mol. Thus the phosphates provide the overall thermodynamic driving force for the carbon fixation.
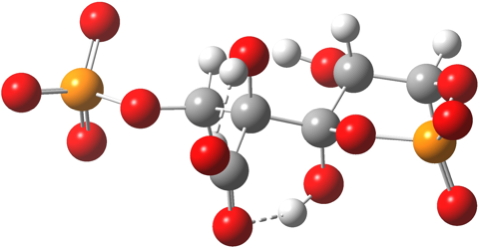
Click for 3D. Cyclic low-energy cyclic chair isomer of 3-keto-2-carboxyarabinitol 1,5-bisphosphate
- Which leaves the role of the enzyme as one of reducing the overall activation barrier. The reaction MUST be enzymatically favoured, since the enzyme also needs to control when the cycle occurs, via a light-sensitive switch. If no enzyme-catalysis were needed, then carbon-fixation would occur in the dark, and consume all available ATP in the process. Inferred purely from the results in the table above, two functions can be listed:
- The enzyme can help increase the effective molarity of the bimolecular reaction between Ribulose-1,5-bisphosphate + CO2. As noted above, increasing the concentration from e.g. 1 atmosphere (0.041M) to 1M reduces ΔG† by 1.9 kcal/mol.
- The most influential role the enzyme could play is to bind the enol form of Ribulose-1,5-bisphosphate preferentially over the keto form. If most of the substrate is bound in this form, that would reduce the overall barrier by 13 kcal/mol, more than enough to enable a room temperature reaction.
- There may of course be many other subtle effects in operation, such as preferential stabilisation of the transition state, which cannot be inferred here without a detailed knowledge of the enzyme. I have deliberately tried to avoid doing that, since I wanted to see what might be concluded purely from the energetics found above.
There is one final step required; a very rapid decomposition of the 3-keto-2-carboxyarabinitol 1,5-bisphosphate (cyclic or not) to produce two molecules of 3-phosphoglycerate. I will leave my computational-energetic analysis and mechanism of that step to another post.
Postscript. An IRC on the full phosphate model took three days to run and has only just finished.[12] The profile is similar to that obtained for the phosphate-free model, with the exception of the IRC feature at -13, where one phosphate group rotates and starts to H-bond to the 3-keto-2-carboxyarabinitol, resulting in a lower energy conformation than that reported above. The energy of this new conformation[13] relative to the starting point (labelled as 0.0 above) is +2.3 kcal/mol (c.f. +11.5 for the previous conformation). The phosphates clearly remain a strong driving force for the reaction. It is quite possible that even more stable forms of this product could be found (by varying where the acidic protons reside) but at least we now know that the product can be more stable than the reactant (by at least -7.3 kcal/mol), which is the important conclusion.


Postscript 1. Yet another lower energy isomer of the product has popped out[14] being -13.1 kcal/mol lower than the initial reactants.
‡I do not describe much molecular biology on this blog, but an urge to rectify this was inspired by a TV program I watched four days ago charting how the pathway chronologically known first as the Calvin, then the Calvin-Benson and now the Calvin-Benson-Bassham cycle for carbon fixation became known (and how it gradually gathered attribution). As a chemist who was trained to try to understand reaction mechanisms, my immediate question (unsurprisingly not addressed at all in the TV program) was: what is the key carbon-carbon bond forming step? Here, I simply wanted initially to answer that one simple question and perhaps the aspect of the relative timing of any C-C bond formation and associated proton transfer. This latter idea in turn was hovering in the background of my mind from association with our previous project in proline-catalysed aldol reactions, where a similar question can be posed and indeed has been answered.[15] The rest of what you see here led directly from trying to answer that initial question. Peter Medawar’s 1963 talk Is the scientific paper a fraud? presented the argument that scientific journal articles give a misleading idea of the actual process of scientific discovery[16]. I hope that perhaps as a blog post, the above does give a little insight into the scientific process I experienced for myself over a period of the last two days (and with conclusions which may of course turn out to be quite wrong).
Acknowledgments
This post has been cross-posted in PDF format at Authorea.
Author
References
- H.S. Rzepa, "Gaussian Job Archive for C5H8O4", 2014. https://doi.org/10.6084/m9.figshare.1004015
- H.S. Rzepa, "Gaussian Job Archive for C5H8O4", 2014. https://doi.org/10.6084/m9.figshare.1004023
- H.S. Rzepa, "Gaussian Job Archive for C5H8O4", 2014. https://doi.org/10.6084/m9.figshare.1004011
- H.S. Rzepa, "Gaussian Job Archive for C5H8O4", 2014. https://doi.org/10.6084/m9.figshare.1004037
- H.S. Rzepa, "Gaussian Job Archive for C5H8O4", 2014. https://doi.org/10.6084/m9.figshare.1004038
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004086
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004066
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004112
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004085
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004111
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004026
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004557
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004614
- H.S. Rzepa, "Gaussian Job Archive for C6H8O13P2(4-)", 2014. https://doi.org/10.6084/m9.figshare.1004778
- A. Armstrong, R.A. Boto, P. Dingwall, J. Contreras-García, M.J. Harvey, N.J. Mason, and H.S. Rzepa, "The Houk–List transition states for organocatalytic mechanisms revisited", Chem. Sci., vol. 5, pp. 2057-2071, 2014. https://doi.org/10.1039/c3sc53416b
- S.M. Howitt, and A.N. Wilson, "Revisiting “Is the scientific paper a fraud?”", EMBO reports, vol. 15, pp. 481-484, 2014. https://doi.org/10.1002/embr.201338302
Tags: 1M solutions, carbon fixation, chair, chemist, energy, free energy, low energy, low energy sink, lower energy conformation, lower energy isomer, Peter Medawar, phosphate
Another way of looking at this reaction is as the reverse of the familiar beta ketoacid decarboxylation.