In a previous post on the topic, I remarked how the regiospecific ethanolysis of propene epoxide[1] could be quickly and simply rationalised by inspecting the localized NBO orbital calculated for either the neutral or the protonated epoxide. This is an application of Hammond’s postulate[[2] in extrapolating the properties of a reactant to its reaction transition state. This approach implies that for acid-catalysed hydrolysis, the fully protonated epoxide is a good model for the subsequent transition state. But is this true? Can this postulate be tested? Here goes.
 Here, I show eight transition state models. As the acid I use CF3CO2H, with methanol as the nucleophile attacking propene epoxide, and I have initially included one additional methanol helping facilitate the proton transfers. Isomeric transition states differ in where the methyl substituent is located (1/2 and 3/4) and in the relative position of the acid and the additional methanol (1/3 and 2/4). In 1/2, the acid is directly protonating the oxygen of the epoxide. In 3/4, it is instead inducing methanol to act as its proxy. Two further transition states 5 and 6 directly replace the CF3CO2H with one (much less acidic) methanol, to test the effect the presence of the acid has on the reaction barriers. Finally, 7 and 8 remove from these models the non-nucleophilic proxy methanol from the ring to test the effect of reducing ring size from 10 to 8.
Here, I show eight transition state models. As the acid I use CF3CO2H, with methanol as the nucleophile attacking propene epoxide, and I have initially included one additional methanol helping facilitate the proton transfers. Isomeric transition states differ in where the methyl substituent is located (1/2 and 3/4) and in the relative position of the acid and the additional methanol (1/3 and 2/4). In 1/2, the acid is directly protonating the oxygen of the epoxide. In 3/4, it is instead inducing methanol to act as its proxy. Two further transition states 5 and 6 directly replace the CF3CO2H with one (much less acidic) methanol, to test the effect the presence of the acid has on the reaction barriers. Finally, 7 and 8 remove from these models the non-nucleophilic proxy methanol from the ring to test the effect of reducing ring size from 10 to 8.
With no catalyst present, we know that the rate of hydrolysis is very slow[1], and that the major product (55%) is the 1-alkoxy-2-propanol, with the 2-alkoxy-1-propanol being the minor component (16%). As acid concentration increases, the amount of the latter eventually exceeds the former. The computed barriers (ωB97XD/6-311G(d,p)SCRF=methanol) for this mode (transition states 5 and 6) are ~29 kcal/mol, which pretty much matches the experimental observation (for ethanol). What does not match is the preference for nucleophilic attack at the least substituted carbon resulting in 1-alkoxy-2-propanol; instead the 2-alkoxy-1-propanol is predicted to have the lower free energy barrier of activation by 1.7 kcal/mol. This will need further investigation in a future post.
| Property | 5, 2-alkoxy-1-propanol | 6, 1-alkoxy-2-propanol. |
| ΔΔG‡, kcal/mol | 0.0 | +1.7 |
| IRC animation | 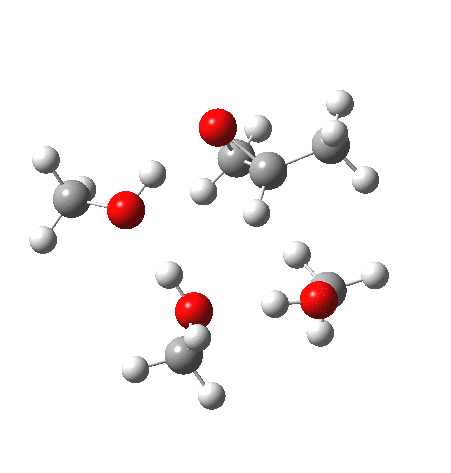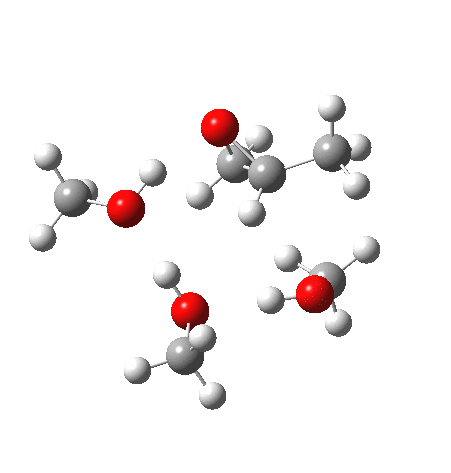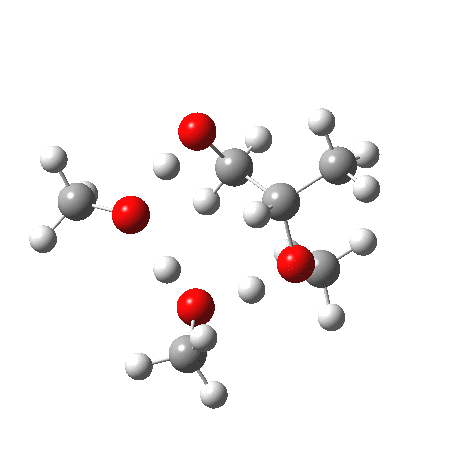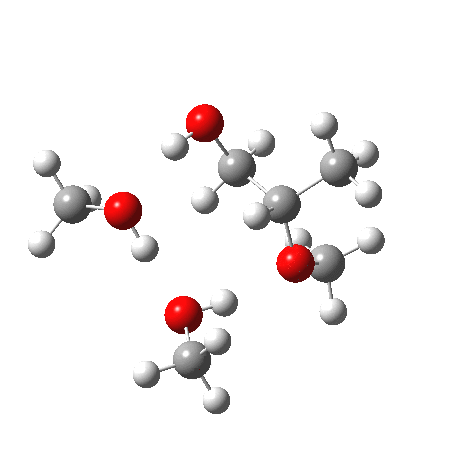 |
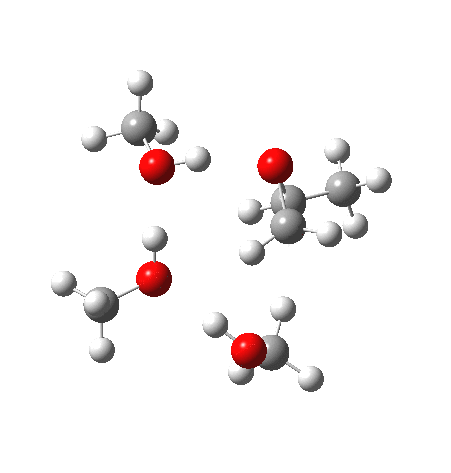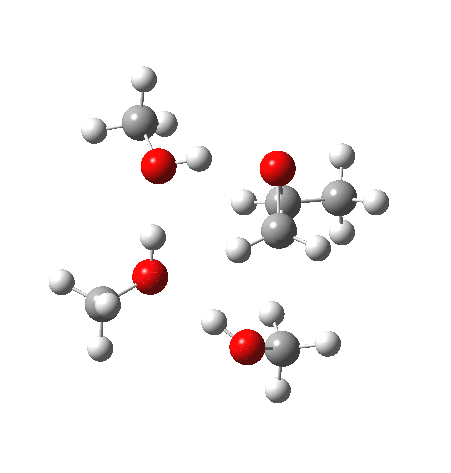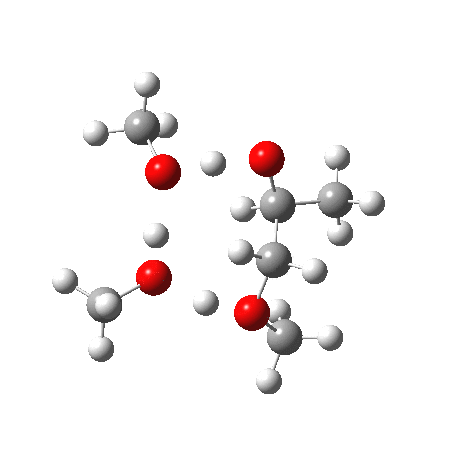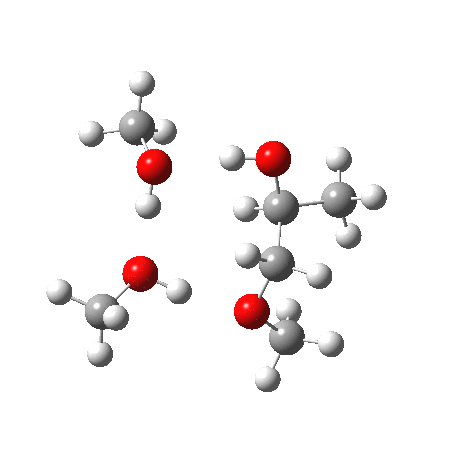 |
| IRC Energy |  |
 |
| IRC Gradients |  |
 |
| IRC | [3] | [4] |
What of the IRCs? Both isomers show an interesting dip in the gradient norms (at~-1.5 for 5 and +1.5 for 6), typical of a “hidden intermediate“. The geometry at this point (below) shows that the erstwhile epoxide bonds are largely formed/cleaved, and this has resulted in a zwitterionic intermediate attempting to form (the nucleophilic oxonium being +ve and the cleaved oxyanion -ve). Such species have no permanence however (not for even one molecular vibration), and are immediately destroyed by three more or less synchronous proton transfers (IRC -2.5 or +3.0). I would add that in many a text-book illustration of this process, this “hidden intermediate” would in fact be exposed as an explicit actual intermediate.
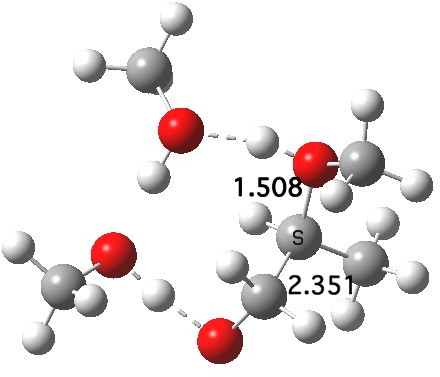
Click for 3D.
What happens when we replace one methanol in the above model with one molecule of trifluoracetic acid, resulting in transition states 1–4 (below).
- The barrier drops dramatically, from ~29 kcal/mol to ~13 kcal/mol. This changes the reaction from a very slow one at room temperatures to a very fast one at room temperatures.
- The IRC now shows an extra “hidden intermediate” before the transition state, as well as one after. The synchronicity of the proton transfers is broken, and now they occur in two distinct stages, one before and one after the transition state. The one before corresponds to protonation of the epoxide oxygen by the trifluoracetic acid, which occurs before the C-O bond is formed/cleaved at the transition state itself. The second hidden intermediate corresponds to the zwitterion arising from the trifluoracetic anion and the oxonium cation located at the original attacking methanol. This is then subjected to proton transfer (IRC ~ -2.5 in both cases) to transfer the proton onto the auxiliary methanol to form what appears to be the final ring-opened neutral product in the presence of methyl oxonium trifluoroacetate.
- So adding a species which can form a stable anion (in other words a strong acid) de-synchronises the reaction. However, all the intermediates are still hidden, and the process is still concerted!
- But, oddly, the predicted preference for 1 is if anything slightly decreased compared to the use of methanol only in the model (i.e. 5/6). This does not seem to correspond to the increased prevalence of 1 in the presence of acid as observed in the experiments.
| Property | 1,2-alkoxy-1-propanol | 2, 1-alkoxy-2-propanol. |
| ΔΔG‡ | 0.0 | +1.4 |
| IRC animation | 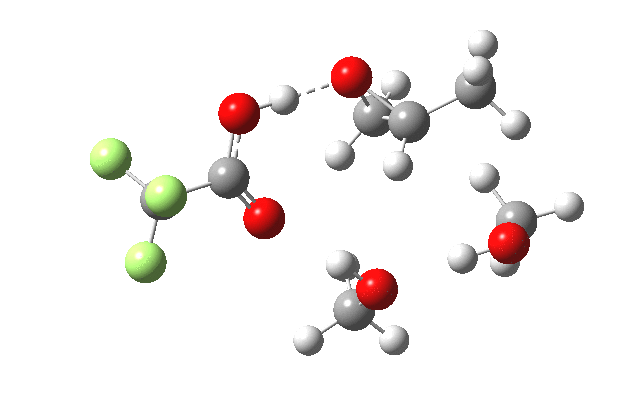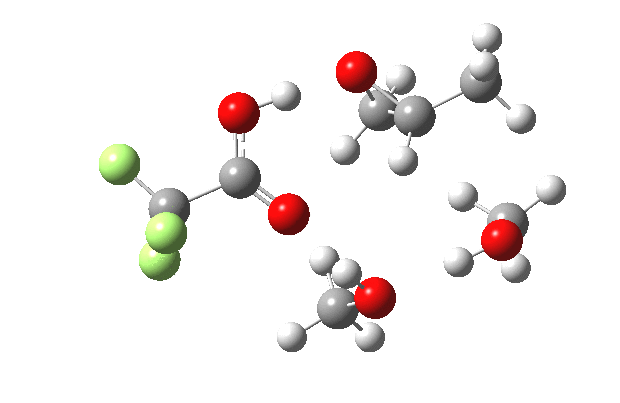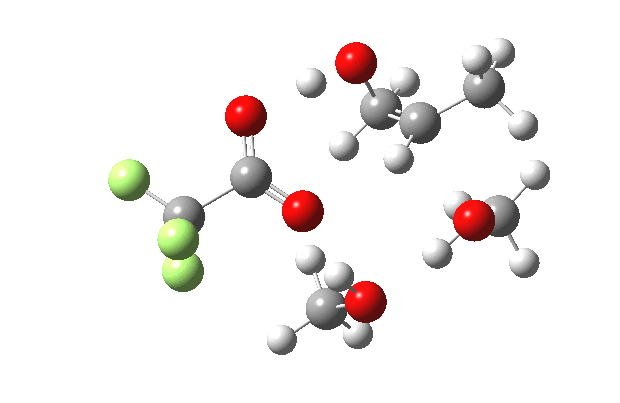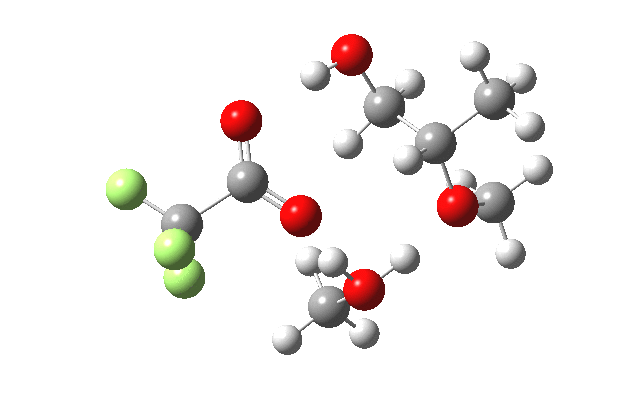 |
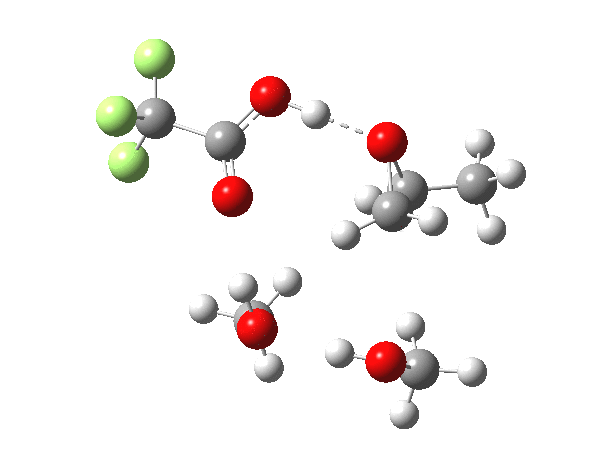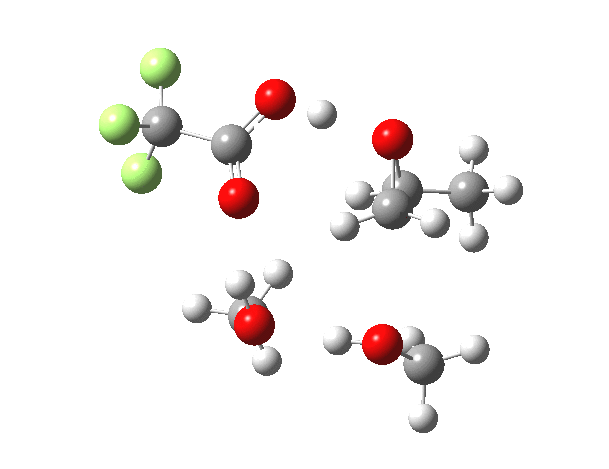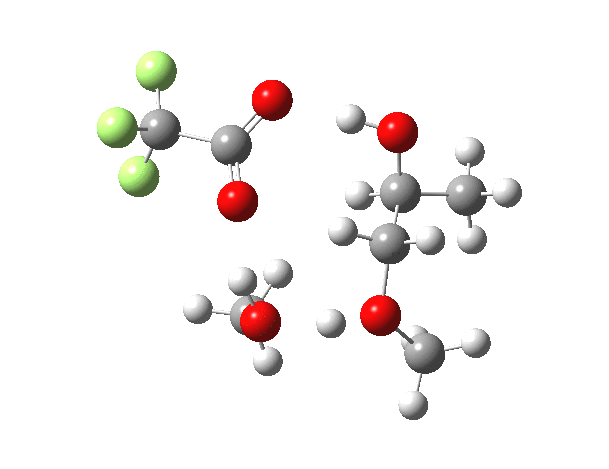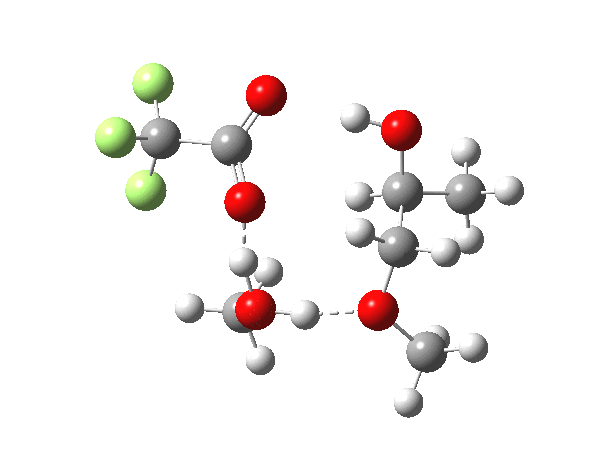 |
| IRC Energy |  |
 |
| IRC Gradients |  |
 |
| IRC | [5] | [6] |
Before moving on to the last models 7/8, I must mention the aspect of where the strong acid is located in the model. If it is located away from the epoxide oxygen, the IRC changes again, now revealing three hidden intermediates.
- The first corresponds to the acid transferring a proton to the non-nucleophilic methanol to form incipient methyl oxonium trifluoracetate
- The second has the methyl oxonium as an acid transferring its proton to the epoxide oxygen.
- Then comes the transition state when the O-C bonds are formed/broken.
- The last hidden intermediate is the oxonium trifluoracetate zwitterion resulting from ring opening, prior to a final proton transfer to reform trifluoroacetic acid.
- This pathway overall in free energy, is about 2.0 kcal/mol higher than the previous one involving direct proton transfer from the acid itself.
| Property | 3, 2-alkoxy-1-propanol | 4, 1-alkoxy-2-propanol. |
| ΔΔG‡ | 1.8 | +3.5 |
| IRC animation | 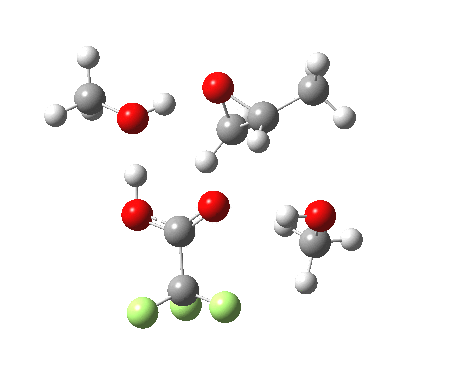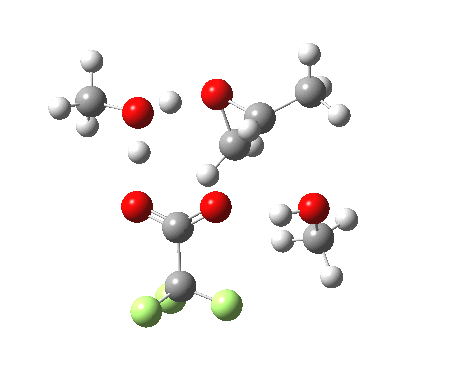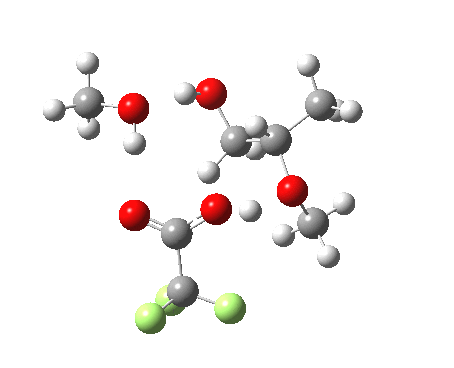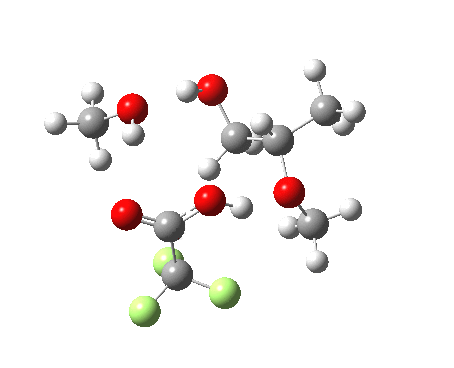 |
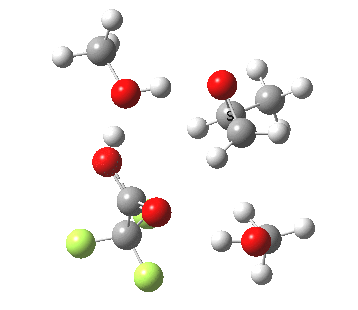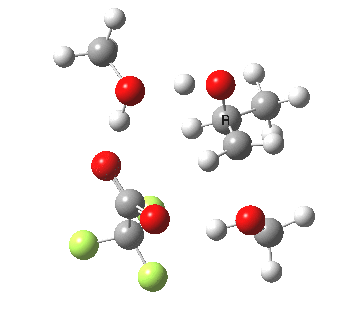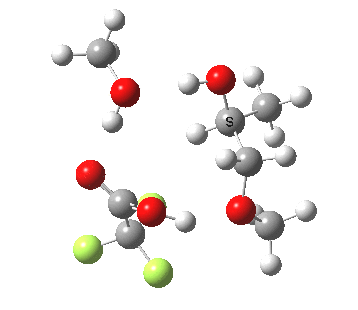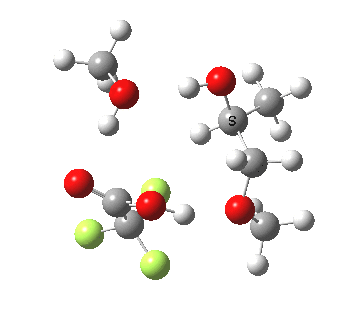 |
| IRC Energy |  |
 |
| IRC Gradients |  |
 |
| IRC | [7] | [8] |
The final model 7/8 tests what happens when that additional methanol is removed from the proton transfer sequence in 1-4. The smaller ring for the transition state induces an increase in the barrier from ~13 to ~20 kcal/mol; this model also naturally “absorbs” an addition methanol to decrease the free energy and mutate into 1-4. The preference for 7 over 8 is increased compared to the other models. The presence of two hidden intermediates in this model is particularly noticeable.
| Property | 7, 2-alkoxy-1-propanol | 8, 1-alkoxy-2-propanol |
| ΔΔG‡ | 0.0 | +3.5 |
| IRC animation | 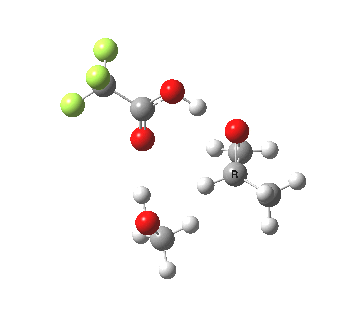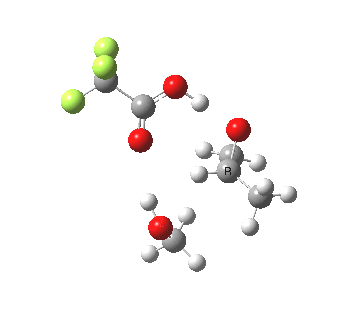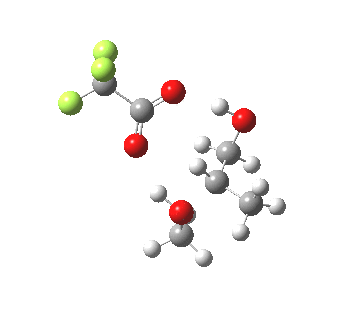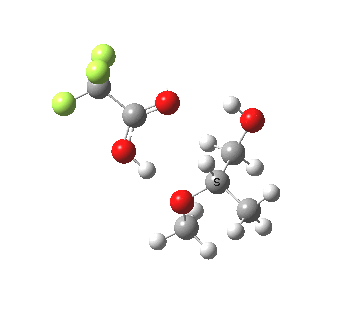 |
 |
| IRC Energy |  |
 |
| IRC Gradients |  |
 |
To answer the question posed at the start of this post, in the IRC explorations above we see that in the presence of trifluoroacetic acid, the transition state is indeed preceded by a proton transfer. This reassures that Hammond’s principle can indeed be applied. The (relative) free energies of the acid catalysed transition state models used here all correctly predict the observed regiochemistry, but we still have to explore the base catalysed route. Watch this space.
Author
References
- H.C. Chitwood, and B.T. Freure, "The Reaction of Propylene Oxide with Alcohols", Journal of the American Chemical Society, vol. 68, pp. 680-683, 1946. https://doi.org/10.1021/ja01208a047
- G.S. Hammond, "A Correlation of Reaction Rates", Journal of the American Chemical Society, vol. 77, pp. 334-338, 1955. https://doi.org/10.1021/ja01607a027
- H.S. Rzepa, "Gaussian Job Archive for C6H18O4", 2013. https://doi.org/10.6084/m9.figshare.694931
- H.S. Rzepa, "Gaussian Job Archive for C6H18O4", 2013. https://doi.org/10.6084/m9.figshare.694918
- H.S. Rzepa, "Gaussian Job Archive for C7H15F3O5", 2013. https://doi.org/10.6084/m9.figshare.694894
- H.S. Rzepa, "Gaussian Job Archive for C7H15F3O5", 2013. https://doi.org/10.6084/m9.figshare.694907
- H.S. Rzepa, "Gaussian Job Archive for C7H15F3O5", 2013. https://doi.org/10.6084/m9.figshare.697508
- https://doi.org/
Tags: CF 3 CO, free energy, good model for the subsequent transition state, lower free energy barrier, Reaction Mechanism
[…] Chemistry with a twist « Hidden intermediates in the (acid catalysed) ring opening of propene epoxide. […]