
The two polymorphs can be easily distinguished by visual inspection; the tetragonal form, 1, grows as blocky, multi-faceted crystals (external form 4/m) with a slight yellow color, while the monoclinic phase, 2, forms absolutely colorless parallelepiped crystals (external form 2/m). In Figure 1, the larger crystal is the tetragonal form 1.
Diffusion of I2 into a chloroform solution of tpp was also found to give the 1:1 complex [9]; however, slow evaporation of this solution gives deeper red crystals of an additional product, identified as tpp· 2I2 [10a]. This latter complex is the sole product of diffusion of I2 into a methylene chloride solution of tpp. When ethanol is used as the solvent a complex mixture of polyiodide salts is formed; in these complexes the tpp molecule is protonated, presumably by water in the solution, at two of the pyridyl nitrogen atoms [10b]. For tpp· I2: IR (nujol, cm-1) 2900 (s), 1461 (s), 1377 (m), 730 (s). Far IR (nujol, cm-1) 634 (w), 547 (m), 406 (w), 172 (s), 100 (m). Anal.: (C24H16N6I2) Calc.: C,44.88%; N, 13.09%; H, 2.51%. Found: C, 47.07%; N, 13.58%; H, 2.56% (the observed deviation in these values is presumably due to loss of I2 from the sample prior to analysis).
The monoclinic to tetragonal phase transformation was accomplished by simply opening a container of the metastable tpp· I2 complex to the atmosphere for approximately one full day. The diffusion of iodine from the complex could be accelerated by warming the solid and/or applying a slight vacuum; this also resulted in a cleaner final product.
Differential scanning calorimetry investigations of tpp were performed on a Perkin-Elmer Series 7 DSC with the DSC software package. A 2.29 mg sample was spread in a thin layer across a high pressure, stainless steel cell and heated at 1.0 š C/min from 240 to 275 š C, then cooled and reheated at the same rate over the same temperature range. Onset calculations were performed by standard methods.
The transition state structure of the tpp solid-state rearrangement was also performed with MOPAC, using a saddle calculation. The transition state geometry was calculated from the crystal structure geometries 1 and 2 and assuming a concerted rotation of the four pyridine rings.
The energy map of dipyridylpyrazine (dpp, Fig. 3) was obtained by modifying the coordinates of 1 by replacing the pyridyl rings on one side of the molecule with hydrogens. The pyrazine-pyridine bond of one of the remaining pyridyl rings was then rotated in 5š increments, beginning with the pyridyl ring co-planar to the pyrazine. The remainder of the molecule was optimized at each step. The data for 73 steps were calculated, giving a complete rotation of the ring. The ³ maintain geometry² keyword was used in order to insure the molecule remained in an optimal view geometry while displaying the energy map with the CAChe Visualizer routine. The screen information was captured with the program Spectator. The animation was then annotated and optimized for presentation with Quicktime applications.
Methylene chloride was found to yield a mixture of the two forms, and chloroform produced large well-formed platey crystals, presumed to be solvated, as they collapsed to a microcrystalline powder of the monoclinic form when removed from the mother liquor.
| Structure |  |  | Tetragonal tpp (in water) | Monoclinic tpp (in water) |
|---|---|---|---|---|
| Compound # | 3 | 4 | 5 | 6 |
| Hš f (kcal/mol) | 173.9 | 176.0 | 150.7 | 150.5 |
Structures 3 and 4 of Table 1 were obtained by performing an energy minimization of the polymorphs 1 and 2 respectively. The geometry minimization resulted in structures only slightly different from the respective starting rotomers. We observe a small, but significant energy difference between the two forms. MOPAC predicts that the monoclinic polymorph will be approximately 2 kcal/mol lower in energy than the tetragonal in the gas phase. If the MOPAC energy minimization is run so as to simulate the environment of a polar solvent (see experimental section for details) the difference in energy between the two forms becomes negligible (structures 5 and 6).
A separate observation is pertinent: When dissolved in chloroform solution, polymorphs 1 and 2 give identical H1-NMR spectra. The rotomers either rapidly interconvert or exist in some intermediate conformation, even on cooling to -78 š C. The lower calculated heat of formation of the monoclinic rotomer, 4, suggests that in non-polar solvents, this conformation is more heavily populated than any ³ tetragonal-type² conformation. Our observation that monoclinic tpp, 2, is preferentially grown from non-polar solvents can be explained simply by energetic considerations. In polar solvents, relative proportion of rotomers is determined by solvent effects which may not be adequately addressed by our model, or by other effects [17]. It is likely however, that the greater stability of the tetragonal packing results in preferential crystallization of 1 in polar solvents.
An interesting feature of the two forms of tpp, which was not explored in any of the previous reports, is the orientation of the pyridine rings with regard to the position of the pyridyl nitrogen atoms. The major difference between the tetragonal and monoclinic forms of tpp involves this aspect of the structure. The tpp molecules in both forms possess crystallographic Ci symmetry, but in the tetragonal form, the pyridyl nitrogen atoms of adjacent pyridine rings lie on the same side of pyrazine ring plane in an endo, exo-conformation, while in the monoclinic form the rings are rotated so that these atoms lie on opposite sides of the pyrazine plane in an endo, endo-conformation. As has been previously noted [8], the tetragonal form of tpp and dpq´ have similar dihedral angles and N--N distances; dpq´ also has a similar ring orientation as both of the pyridine nitrogen atoms are on the same side of the quinoxaline ring plane (Fig. S3)). The monoclinic form of tpp, dpp and dpq also have similar dihedral angles and N--N distances, and all have the nitrogen atoms of adjacent pyridine rings on opposite sides of the diazine ring plane (Fig. S4).

Our single crystal diffraction studies show a lower average thermal motion of the tetragonal rotomer (0.042 Å 2) as compared the monoclinic rotomer (0.050 Å 2) at room temperature. The tetragonal form is therefore the lower energy conformation at room temperature, but the relative stabilities reverse upon heating. At about 258 š C, there is sufficient energy available to the crystal that defects in the lattice are formed [14]. These disruptions are rapidly propagated through the solid, allowing rotation of the pyrazine rings and the reordering of the lattice into a monoclinic space group.
 While the important motions available to tpp are limited to rotations
about the four pyridine-pyrazine bonds, the potential energy surface is complex
and pocketed with numerous local minima. We performed a MOPAC saddle calculation
to give us a better idea of the transition state between the two forms of tpp. In
order to do this, we assumed that the rotation of the four rings was linear and
concerted, a feat that is extremely unlikely, particularly in a solid state
process where indirect trajectories may occur due to mechanical stress induced by
the matrix [15]. Despite these caveats, the ³
transition state² (Structure 7) proves to be a
convenient model for analyzing the reaction pathways discussed below. This
structure has a center of inversion with two pyridyl rings parallel and two
perpendicular to the pyrazine ring.
While the important motions available to tpp are limited to rotations
about the four pyridine-pyrazine bonds, the potential energy surface is complex
and pocketed with numerous local minima. We performed a MOPAC saddle calculation
to give us a better idea of the transition state between the two forms of tpp. In
order to do this, we assumed that the rotation of the four rings was linear and
concerted, a feat that is extremely unlikely, particularly in a solid state
process where indirect trajectories may occur due to mechanical stress induced by
the matrix [15]. Despite these caveats, the ³
transition state² (Structure 7) proves to be a
convenient model for analyzing the reaction pathways discussed below. This
structure has a center of inversion with two pyridyl rings parallel and two
perpendicular to the pyrazine ring.If we make the assumption that the pyridyl rings on opposite sides of the pyrazine interact only minimally, then the primary features of the ring rotation can be modeled by dpp. We have previously characterized dpp in the solid state, and have found its pyridyl rings to be oriented much like those of 2 [12]. We will refer to this as ³ monoclinic² form of dpp and will also discuss the hypothetical ³ tetragonal² rotomer. These terms serve to highlight the relationship between these structures and the tpp polymorphs and do not have any relation to the actual crystal system observed for dpp [12].
Figure 3 shows the energy map resulting from a MOPAC dihedral drive calculation. One of the pyridyl rings was rotated in 5 degree increments about the pyridyl-pyrazine bond, beginning from a co-planar pyridyl-pyrazine orientation. The rest of the molecule was energy minimized at each step. Figure 3 plots heats of formation vs dihedral angle.
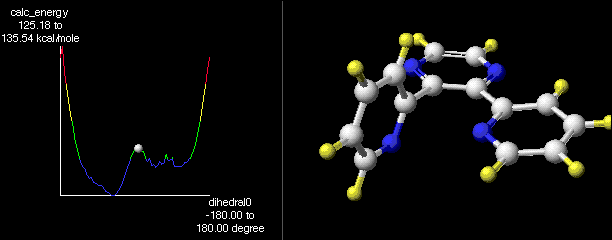
Three low energy wells are observed in this experiment. As expected, the lowest of these (by about 2 kcal/mol) corresponds to the ³ monoclinic² conformer while the other two are enantiomers of the ³ tetragonal² conformer. There is a very small energy barrier (about 0.8 kcal/mol) between these conformers. The activation enthalpy for conversion from the ³ tetragonal² to the ³ monoclinic² rotomer is also quite small, about 1.2 kcal/mol.
It is clear from Figure 3, that the highest energy conformations are those with the controlled pyridyl ring co-planar to the pyrazine. Of the two orientations having this feature, the one in which the nitrogen of the controlled pyridyl ring is turned towards the other pyridyl ring is substantially lower in energy. This orientation looks very much like that proposed in 7. By rotating one pyridyl ring into co-planarity, the other is forced into a nearly perpendicular orientation. From this, we might also conclude that anything which encourages one ring to twist towards a perpendicular orientation relative to the pyrazine might also facilitate interconversion of ³ tetragonal² and ³ monoclinic² orientations.
Although crystals suitable for a single crystal structure determination of the tpp· I2 complex have not been isolated, the elemental analysis (corrected for I2 loss, which is known to occur) agrees with our interpretation. Additionally, the Far-IR spectrum of this compound exhibits a strong signal at 172 cm-1, typical of an I-I stretching frequency for pyridine I2 complexes [16]. Thermal gravimetric analysis of this sample showed a single mass loss with an onset of 117.5 š C. In this event, 40% of the total mass was lost (calc. 39.5%).
The addition of I2 to tpp has profound structural consequences. Table 2 gives the calculated heats of formation for five possible complexes between tpp and I2.
| Structure | Structure 1+ I2 on endo ring | Structure 1+ I2 on exo ring | Structure 2+ I2 on a pyridyl ring | Structure 1+ I2 on the pyrazine ring | Structure 2+ I2 on the pyrazine ring |
|---|---|---|---|---|---|
| Compound # | 8 | 9 | 10 | 11 | 12 |
| Hš f (kcal/mol) | 189.8 | 188.6 | 195.2 | 197.7 | 196.5 |
| Hš rxn (kcal/mol) | -6.95 | -8.15 | +0.56 | +0.91 | +1.83 |
The tetragonal form has two different types of pyridyl rings, which will be labelled as the endo (pyridine nitrogen pointed away from the pyrazine nitrogen) and exo (pointing towards the pyrazine nitrogen) rings. Addition of I2 to either ring is predicted to be an exothermic process by about 7.5 kcal/mol (8 and 9) with coordination at the exo ring appearing to be slightly favored. In each case, steric interactions force the ring bearing the I2 into a nearly perpendicular orientation to the central pyrazine ring. Since perpendicular orientation of a pyridyl ring is required for a low energy rotation of an adjacent ring, the I2 complex would be expected to convert from one rotomer to another with relative ease. Interestingly, as the coordinated ring moves more towards a perpendicular geometry, the difference between the ³ endo² and ³ exo² coordinated forms becomes less significant. Structures 8 and 9 are essentially equivalent.
In addition to changing the geometry of the two rings on the side of the molecule coordinated to I2, the opposite side of the molecule is also affected, though to a lesser degree. For example, on coordination of I2 to an exo ring, both the coordinated and non-coordinated exo rings move into geometries closer to perpendicular to the pyrazine ring, while both endo rings become more parallel.
MOPAC calculations gave a very different result when an I2 complex is generated from the monoclinic tpp structure 2. The optimized structure is a local minima (10) that is perturbed only slightly from the starting compound. Enthalpy calculations predict that this structure is significantly higher in energy than the conformers generated from the tetragonal form and the formation of such a structure was found to be endothermic by about 0.56 kcal.
It is also conceivable that the iodine adds to one of the pyrazine nitrogens. While pyrazine· I2 complexes are known [6,10a], the pyrazine moiety does not appear to seriously compete with pyridine for I2. Structures 11 and 12 show that pyrazine· I2 coordination starting from either rotomer results in a high energy species. These will not be considered further.
We suggest that the actual solid state structure of the tpp· I2 complex is similar to that of 8 and 9 with some allowances made for packing energetics. This is supported by the TGA data and the FIR data, both of which are consistent with related azoaromatic charge-transfer complexes [7,10a]. Loss of I2 from this species would be expected to proceed with minimum disruption of the tpp ring orientation and would lead therefore, to the observed product, 1.
{kind=link}
{kind=link}
{kind=link}