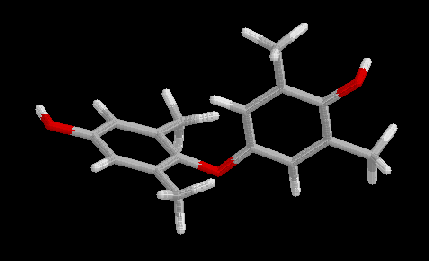
The polymer was represented by the following compound:
Based on previous ground state studies of the possible conformations of the central ether bond [2] the terminal OH-groups were fixed to reflect the minimum conformation of the ether bond in the polymer.
All calculations were carried out with the program package VAMP 5.5 [5] using the standard AM1 method. The ground state structures of the different conformations were fully optimized with the UHF-AM1 method, using the eigenvector following method for minima [6] and the NS01A method |7] for transition states. The excited state geometries were not optimized.
The fact that in both intramolecular mechanisms studied, biradicaloid intermediates or products are formed, aggravated the calculation of excited state energies. Thus CI-calculations should be carried out using the calculated UHF wavefunction. Unfortunately, this method was not implemented in the used program and even larger CI calculations were not able to reproduce the UHF ground state potential energies satisfactorily. Therefore the Si and Ti energies were calculated using the following equations:
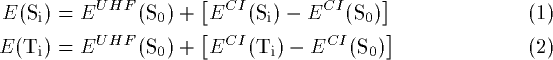
The CI calculations included all singly and doubly excited configurations which can be constructed from the 5 highest occupied and the 5 lowest unoccupied orbitals leading to a total of 876 configurations. A direct calculation of the T1 states was unfortunately not possible due to the large spin contamination of the UHF wavefunction.
 Contents
Contents  Introduction -
Results and Discussion
Introduction -
Results and Discussion
