 Molecular mechanics calculations were done for
ent-13-epi-manoyl oxides with hydroxylations at different positions
of the skeleton (compounds 1-18).
According to the numerical results obtained, the conformational behavior of the
products and their geometric tendencies were established. The theoretical
coupling constants were calculated, giving results consistent with the
experimental data. Moreover, puckering parameters of the different rings of the
molecule were determined, and these values made conformational analysis
possible.
Molecular mechanics calculations were done for
ent-13-epi-manoyl oxides with hydroxylations at different positions
of the skeleton (compounds 1-18).
According to the numerical results obtained, the conformational behavior of the
products and their geometric tendencies were established. The theoretical
coupling constants were calculated, giving results consistent with the
experimental data. Moreover, puckering parameters of the different rings of the
molecule were determined, and these values made conformational analysis
possible.

Molecular mechanics calculations were done for
compounds 1-18. These compounds are
tricyclic diterpenes belonging to the enantio series (ent-), with
ent-13-epi-manoyl oxide structure.
Compound 1 is the unfunctionalized skeleton
of ent-13-epi-manoyl oxide. Compounds 2-15 are
ent-13-epi-manoyl oxides with different hydroxyl groups at positions C-3,
C-6, C-7, C-11 and/or C-12 with different ent-alpha or ent-beta
arrangements. Compounds 16-18 are mono- or diketones at positions C-3 and
C-12.
These compounds were chosen for molecular mechanics
calculations for the following reasons:
 a) they are related to the experimental work
carried out by our research group, since they arise from natural, chemical or
microbiological sources;
a) they are related to the experimental work
carried out by our research group, since they arise from natural, chemical or
microbiological sources;
b) experimental coupling constants are known;
c) the calculated theoretical coupling constants
are helpful in assigning experimental coupling constants;
d) all the structures have three fused highly
rigid rings, making investigations of their conformational behavior of
interest.
Molecular mechanics methodology [1] was used throughout the study. Molecular mechanics
modelling and conformational analyses of compounds
1-18 were done with the PC-MODEL program [2]. Energy
minimization was done with the program using an MMX force field, which was a
modification of the MM2 (QCPE-395, 1977) [3] and MMP1
(QCPE-318) programs [4].
Conformational analysis of rings and hydroxyl
orientation in structures 1-18 was done with the RANDOMIZE and MULTOR
options of the PC-MODEL program. This conformational analysis provides the lowest
energy conformations. From these conformations, steric energies and theoretical
coupling constants were calculated using the PMR option of the PC-MODEL program.
This option is based on an empirical generalization of the Karplus equation [5].
Cremer-Pople [6] ring
puckering parameters were calculated for the different rings of the most stable
conformations for compounds 1-5, 8, 16-18, using the CONPUC program [7,8].
The PLUTO drawings [9] of
the most stable conformations of compounds 1-4,
5-8, 9-14 and
15-18 (including the structures of these
compounds with the C ring in the boat conformation) were done.
The energy differences for compounds 1-5, 8
and 18 are approximately of 3-7 kcal/mol in favor of those conformations
with a C ring in a chair conformation. Therefore, we conclude that products
1-18 exist mainly in only one ring conformation, with the A, B and C rings
very close to the chair conformations.
The unfunctionalized ent-13-epi-manoyl oxide
skeleton has a steric energy of 47.9 kcal/mol. Compounds 2
(ent-3beta-hydroxy) and 3 (ent-12alpha-hydroxy) are mono-hydroxylated
derivatives. Compounds 4, 5, 6 and 7 have two hydroxyl groups;
compounds 4 and 5 are epimers at C-12 and compounds 6 and 7 are
epimers at C-6. In both cases the equatorial arrangement (ent-6a or ent-12b) is
energetically favorable in approximately 0.5 kcal/mol. Products 8-15 are
trihydroxylated compounds. Some of them (9 and 12) have small
steric energy values (47.3 and 46.5 kcal/mol, respectively). Both products
9 and 12 have the same functionalization at C-3, C-7 and C-12,
although opposite arrangements at C-7 and C-12. Compounds 10 and 13
are epimers at C-12 and the equatorial arrangement is again favorable (0.3
kcal/mol). Compounds 16 and 17, epimers at C-1, have a carbonyl
group at C-3. Diketone 18 is abnormally stable since its steric energy is
46.1 kcal/mol.
Cremer-Pople ring puckering parameters were
calculated for the different rings of the most stable conformations for compounds
1-5, 8, 16-18.The B ring for all products (1-18), independently
of the C ring conformation, has a chair conformation. The C ring for all
the compounds presents a slightly distorted chair conformation.
The experimental data for products 2-18
indicate that the A, B and C rings are preferably in a chair conformation,
with good concordance between the theoretical and experimental values of the
coupling constants for the protons on C-2 and the geminal proton to the
equatorial hydroxyl group on C-3 (for A ring), coupling constants for protons on
C-6 and geminal proton to the axial or equatorial hydroxyl group on C-7 (for B
ring) and coupling constants between H-9 and H-11 (for C ring).

 Abstract
Abstract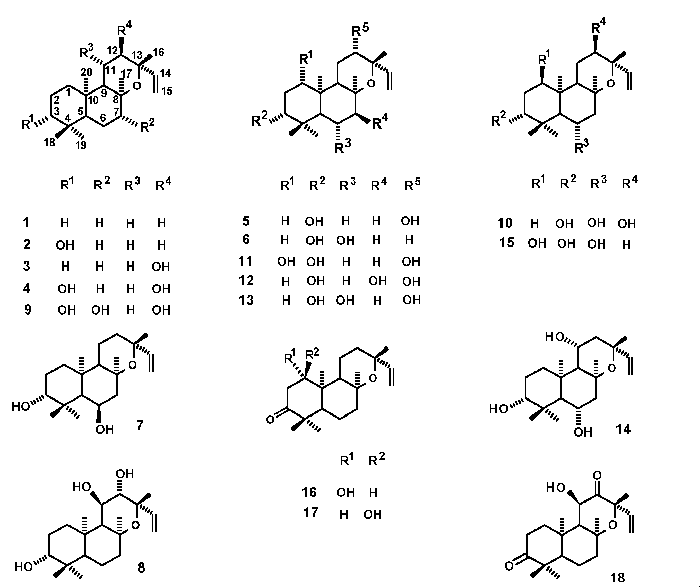{kind=link}