A Test of the Single
Conformation Hypothesis in the Analysis of NOESY Distance Data for Small
Molecules
James P. Snyder,*
Neysa Nevins, Daniel Cicero and Dennis Liotta
Emerson Center for Scientific Computation, Department of Chemistry, Emory
University, 1515 Pierce Drive, Atlanta, GA 30322
Abstract
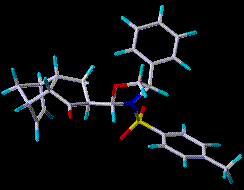 Tricyclic ketone 1 has been subjected by Reggelin and coworkers to a
careful NOESY analysis in CDCl3. Data processing by restrained molecular dynamics (MD) led to the proposition of a single
conformation in solution [1]. The present work examines the validity
of this interpretation.
Specifically, the conformational profile of the endo isomer of 1
was examined by a 1000 step Monte Carlo search with MM2* and the GB/SA CHCl3
solvent continuum model in MacroModel [2]. The structures of the 58 unique and fully
optimized conformations and the NMR-derived geometric variables were together subjected to a NAMFIS
analysis (NMR Analysis of Molecular Flexibility in Solution) [3]. Ideally, the
procedure deconvolutes the thermally averaged NMR data into a small family of
conformations that optimally represents the J-derived torsions and the
nOe-derived distances.
We conclude that tricyclic ketone 1 is not well-characterized by a
single conformation in CDCl3 solution, but by a set of rapidly equilibrating
conformers that produce a deceptively averaged NMR spectrum. It would appear
that the previously suggested structure is a virtual conformation obligated to
compress multi-conformer features within a single 3-D construct.
Tricyclic ketone 1 has been subjected by Reggelin and coworkers to a
careful NOESY analysis in CDCl3. Data processing by restrained molecular dynamics (MD) led to the proposition of a single
conformation in solution [1]. The present work examines the validity
of this interpretation.
Specifically, the conformational profile of the endo isomer of 1
was examined by a 1000 step Monte Carlo search with MM2* and the GB/SA CHCl3
solvent continuum model in MacroModel [2]. The structures of the 58 unique and fully
optimized conformations and the NMR-derived geometric variables were together subjected to a NAMFIS
analysis (NMR Analysis of Molecular Flexibility in Solution) [3]. Ideally, the
procedure deconvolutes the thermally averaged NMR data into a small family of
conformations that optimally represents the J-derived torsions and the
nOe-derived distances.
We conclude that tricyclic ketone 1 is not well-characterized by a
single conformation in CDCl3 solution, but by a set of rapidly equilibrating
conformers that produce a deceptively averaged NMR spectrum. It would appear
that the previously suggested structure is a virtual conformation obligated to
compress multi-conformer features within a single 3-D construct.
- Introduction
- Methods
- Results
- Conclusions
- References
Introduction
Multi-dimensional NMR has become a powerful complement to X-ray
crystallography for the determination of molecular structure in solution.
Among others, the three-dimensional architectures of proteins, oligonucleotides
and macromolecular complexes have been convincingly established in aqueous
media. [4] The methodology generally involves the measurement of a large
number of geometrically determined variables such as nOe's and coupling
constants (J's) followed by a search of the target structure in 3-space
constrained by the same variables. For example, starting from a number of
points, restrained molecular dynamics (MD) routinely leads to a common, internally
consistent 3-D structure. [5] The tactics are successful, in part, because the
macromolecules in question are relatively conformationally immobile. Apart
from small subsets of disordered loops, the molecules present essentially a
single target conformation.
Peptides or small flexible organic molecules show a fundamentally different
conformational profile in solution across a range of temperatures. That is,
the compounds are usually characterized by families of rapidly equilibrating
conformations characterized by an average NMR spectrum. While there are a few
approaches to deconvoluting the average, [6] it has become popular to apply the
macromolecular techniques to small molecules in solvent under the assumption of
a single important conformation. Structures reasonably consistent with NOESY,
ROESY and coupling constant data are thereby derived and offered as a
meaningful solution to the conformational problem. [7,8,9]
The present contribution examines this proposition for tricyclic ketone
1, an enantiomerically pure diastereomer synthesized by Diels-Alder cycloaddition in a study of asymmetric induction.[10]
Compound 1 has been subjected by Reggelin and coworkers to a careful
NOESY analysis in CDCl3 leading to 50 apparently well-determined intramolecular
distances and one key J(H1-H6) = 2.9 Hz. [1] Restrained MD and the assumption of
a strongly preferred conformation yielded a diastereomeric
configuration and conformation similar to that for a closely related structure
derived by X-ray crystallography. [11]
Methods
In the present work, conformational profiles of the endo and exo isomers of
1 were obtained by 1000 step Monte Carlo searches with the MM2* force
field and the GB/SA CHCl3 solvent continuum model in MacroModel. [2]
A total of 37 and 58 fully optimized conformations within 10 kcal/mol of the
respective global minima resulted. The fact that the global minima were found
15 and 13 times, respectively, provides a high measure of confidence that a
thorough search of the low energy regions of conformation space has been
achieved.
A NAMFIS analysis was carried out for each of these data sets with the nOe determined distances and J(H1-H6).The
recently disclosed method (NMR Analysis of Molecular
Flexibility in Solution) [3] combines empirical and
modeled structural variables by a non-linear least squares procedure to yield a
"best fit" solution to the geometrically transformed 2-D NMR spectrum (or
spectra). Goodness-of-fit is expressed as the sum of square differences (SSD)
between measured and modeled variables. In practice, the procedure operates on a "complete" set of molecular conformations and
deconvolutes the thermally averaged NMR variables into a small family of
conformations that optimally represents the J-derived torsions and nOe-derived
distances. The population of conformers (set of molar fractions) that best
fits both the experimental NMR data and the associated errors is considered a "feasible solution". In the present case a separate error analysis has not been performed for each NMR distance and torsion. Instead, the following error estimates are assigned to the nOe distance categories below:
error nOe distances (x in Å)
+/- 0.1 x < 2.5
0.2 2.5 <= x < 3.0
0.3 3.0 <= x < 3.5
0.4 3.5 <= x < 6.0
The result of the fitting exercise is thus termed a "best solution". Roughly speaking, the outcome is 80% probable at the 95% confidence level.
Results
The exo dataset yielded a relatively poor four-conformation fit to the NMR
data with SSD = 45. The outcome is consistent with the previous
nOe/restrained MD examination which ruled out the exo isomer as the product of
the Diels-Alder reaction. [1] By contrast, the endo dataset delivered a
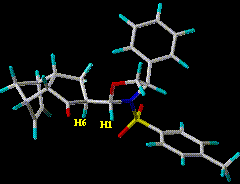
four-conformation fit with SSD = 9 and J(H1-H6) = 2.6 Hz (Jexp = 2.9 Hz). Most of this error is due to deviations in calculated and measured nOe distances involving the meta protons on the phenyl subunit.
Tables 1 and 2
display the predicted percentages of each conformer in
the endo and exo sets, respectively, as well as the values of the
J(H1-H6)-derived torsion. For comparison, the single restrained MD conformation
of 1 was reconstituted from data in the Reggelin work by reoptimizing
the structure (MM2*) under the constraint that both the torsional angles around C1-C6 and the
intramolecular distances conform to the reported MD values (SSD = 3 when fit
to MD values). However, NAMFIS fitting of the MD structure to the NMR data led to the unusually
high SSD = 31 and J(H1-H6) = 3.5 Hz. The MM2* MD-structure was subsequently
reoptimized without constraints leading to an energy drop of 16.4 kcal/mol. The structure is clearly distant from the nearest local minimum on the MM2* potential energy surface.
The MD structure obtained above (white in figure) was
compared with the "best fit" family of endo conformers. Torsions describing the
two rings (oxazolidine, cyclopentanone), the C1-C6 dihedral, and the torsions
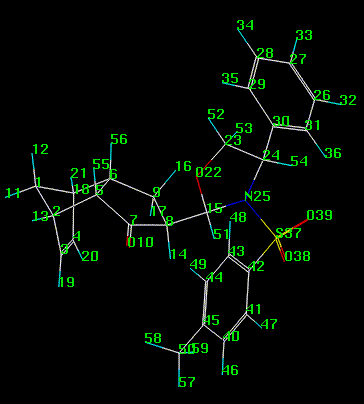
about N-S, S-Ctol, and C-Cph are listed in Tables 3 & 4 and Tables 5 & 6. Figure 3 illustrates the atomic
numbering. The MD conformer
is distinct from each of the conformers in the NAMFIS population. For comparison, the values
for an average structure of the NAMFIS conformers (weighted by mole fraction)
is also included in Tables 3 - 6. The average, a "virtual" conformer, is actually closer to
the MD structure than any individual optimized conformer.
Conclusions
We conclude that tricyclic ketone 1 is not well-characterized by a
single conformation in CDCl3 solution, but by a set of rapidly equilibrating
conformers that produce a deceptively averaged NMR spectrum. It would appear
that the previously suggested structure is a virtual conformation which combines
multi-conformer features in a single geometry. As such, the
probability of its existence on the conformational surface of the endo
Diels-Alder adduct is negligible. In general, the presumption of single or
strongly preferred conformation for a small molecule characterized by easily
rotated bonds involves both a risk and a deep-seated assumption. The assertion is best tested by independent means
prior to mapping NMR distances and torsions onto a single aggregate of atoms.
References
[1] Reggelin, M.; Hoffmann, H.; Kock, M.; Mierke, D.F. J. Am. Chem.
Soc. 1992,114, 3272.
[2] Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.;
Caufield, C.; Chang, G.; Hendrickson, T.; Still W.C. J. Comp. Chem.
1990, 4, 440.
[3] Cicero, D.O.; Barbato, G.; Bazzo, R. J. Am. Chem. Soc.
1995,117, 1027.
[4] Wutrich, K.; NMR of Proteins and Nucleic Acids; Wiley: New York, 1986.
[5] Brunger, A. T.; Clore, G. M.; Gronenborn, A. M.; Karplus, M; Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 3801; Kuszewski, J.; Nilges, M.; Brunger, A. T. J. Biomol. NMR, 1992, 2, 33; Powers, R; Garrett, D. S.; March, C. J.; Frieden, E. A.; Gronenborn, A. M.; Clore, G. M. Biochemistry, 1993, 32, 6744.
[6] Landis, C; Allured, V. S. J. Am. Chem. Soc., 1991, 113, 9493; Bruschweiler, R.; Blackledge, M.; Ernst, R. R. J. Biomol. NMR, 1991, 1, 3; Nikiforovich, G. V.; Prakash, O.; Gehrig, C. A.; Hruby, V. J. J. J. Am. Chem. Soc., 1993, 115, 3399.
[7] Cachau, R. E.; Gussio, R.; Beutler, J. A.; Chmurny, G. N.; Hilton, B. D.; Muschik, G. M.; Erickson, J. W. Supercomputer Applic. and High Performance Comput. 1994,8, 24.
[8] Paloma, L. G.; Guy, R. K.; Wrasidio, W.; Nicolaou, K. C. Chemistry and Biology 1994, 1, 107.
[9] Walker, S.; Valentine, K. G.; Kahne, D. J. Am. Chem. Soc., 1990, 112, 6428; Walker, S.; Yang, D.; Kahne, D. J. Am. Chem. Soc., 1991, 113, 4716; Walker, S.; Murnick, J.; Kahne, D. J. Am. Chem. Soc., 1993, 115, 7954.
[10] Hoffmann, H.; Bolte, M.; Berger, B.; Hoppe, D. Tetrahedron Lett. 1993, 6537.
[11] Citations 18 in reference [1].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}