
Before this work could be applied to the preparation of anthracenones analogous to the A-B-C core of olivomycin A, it was necessary to establish that the electrocyclic reaction sequence would operate with naphtho[b]cyclobutenes. This was by no means guaranteed, since the thermal transformation of a naphthocyclobutene 3 to an o-quinonedimethide 4 involves the complete disruption of a naphthalene aromatic system, and the activation barrier can therefore be expected to be higher than that leading to the monocyclic systems 2. This effect is reflected in the higher temperature required to convert 3 into 4 as compared to 5 into 6 [4], the problems encountered in attempting to generate the thia system 7 as compared to 8 [5], and the slower rate of formation of 9 as compared to 10, from the respective nitroazide precursors [6].

In order to address this issue, we prepared the naphthocyclobutenes 12 and 13 and studied their thermal electrocyclic ring opening reactions (Scheme 2). We prepared the ketone 11 from 3-amino-2-naphthoic acid using a conventional aryne [2 + 2] cycloaddition method [7]; there is an alternative route to 11 via naphtho[b]cyclopropene [8]. The addition of the Grignard reagent vinylmagnesium bromide to 11 in THF at -10 proceeded in good yield, and gave the alcohol 12 as colourless needles, m.p. 95-96 Methylation gave the ether 13 as a colourless oil.
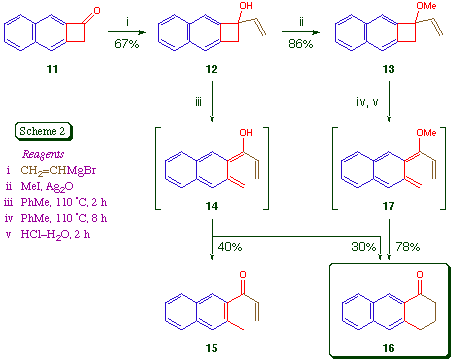
Thermolysis of the carbinol 12 in toluene solution under nitrogen resulted in its complete disappearance in two hours. The major products were the ring-opened enone 15 (40%) and the known [9] anthracenone 16 (30%), which were isolated by chromatography. The formation of a significant amount of 15, which is formally derived from 14 via a 1,5-hydrogen shift, has no analogy in the monocyclic series [3], and is presumed to be a manifestation of the higher energy (and greater diradical character) of dienes of the naphtho[b] fused series. It certainly does not seem likely that the ground-state Z-isomer of 14 is the major precursor of 15 [3,10]. As expected, the formation of 15 was suppressed by the simple expedient of O-methylation. When the ether 13 was heated at 110 for eight hours, acid hydrolysis (4 M HCl, 2 h) of the intermediate enol ether gave the desired anthracenone 16 cleanly and in good yield.
We are currently seeking new routes to highly oxygenated naphtho[b]cyclobutenones.
REFERENCES
1 For a review, see Remers, W.A. The Chemistry of Antitumour Antibiotics; Volume 1; Wiley: New York, 1979, p. 133 et seq.
2 For leading references, see Datta, S.C.; Franck, R.W.; Noire, P.D. J. Org. Chem. 1984, 49, 2785; Roush, W.R.; Michaelides, M.R.; Tai, D.F.; Lesur, B.M.; Chong, W.K.M.; Harris, D.J. J. Am. Chem. Soc. 1989, 111, 2984; Sebesta, D.P.; Roush, W.R. J. Org. Chem. 1992, 57, 4799; Toshima, K.; Nozaki, Y.; Nakata, M.; Tatsuta, K.; Kinoshita, M. Tetrahedron Lett. 1993, 34, 5761; Roush, W.R.; Lin, X.-F. Tetrahedron Lett. 1993, 34, 6829.
3 Hickman, D.N.; Wallace, T.W.; Wardleworth, J.M. Tetrahedron Lett. 1991, 32, 819.
4 Cava, M.P. J. Org. Chem. 1962, 27, 755.
5 Mayer, A.; Meier, H. Tetrahedron Lett. 1994, 35, 2161.
6 Dyall, L.K.; Ferguson, J. Aust. J. Chem. 1994, 47, 0000.
7 Abou-Teim, O.; Goodland, M.C.; McOmie, J.F.W. J. Chem. Soc., Perkin Trans. I 1983, 2659.
8 Durucasu, I.; Saraçoglu, N.; Balci, M. Tetrahedron Lett. 1991, 32, 7097. See also Müller, P.; Bernardinelli, G.; Jacquier, Y.; Ricca, A. Helv. Chim. Acta 1989, 72, 1618.
9 Agranat, I.; Shih, Y.-S. Synthesis 1974, 865; Boger, D.L.; Zhou, J. J. Org. Chem. 1993, 58, 3018.
10 For the basis of this assertion, see Arnold, B.J.; Sammes, P.G.; Wallace, T.W. J. Chem. Soc., Perkin Trans. I 1974, 415. See also Moss, R.J.; White, R.O.; Rickborn, B. J. Org. Chem. 1985, 50, 5132; Rondan, N.G.; Houk, K.N. J. Am. Chem. Soc. 1985, 107, 2099.