
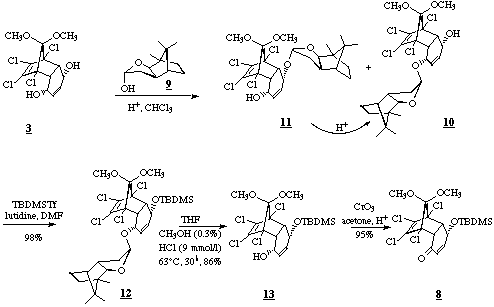
According to Marchand's procedure [4], meso-diol 3 was prepared from benzoquinone and 5,5-dimethoxy-1,2,3,4-tetrachlorocyclopentadiene in high yields.Treatment of diol 3 with half an equivalent of the dimeric lactol 4 [5] under acidic conditions led to the diastereomeric monoacetals 5 and 6 (5 : 6) in 87% yield. The low diastereoselectivity is of minor importance, because acidic equilibration of the two products is possible. After separation by column chromatography on silicagel (deactivated with triethylamine), silylation of 6 (t-butyldimethylsilyltriflate, 2,6-lutidine in DMF, 0deg.C, 2h,100%) leads to 7 (X-Ray). Treatment with Jones reagent results in hydrolysis of the acetal and oxidation to the enone 8. The chiral auxiliary can be recycled as the lacton, which can easily be reduced to the lactol.
An improved variant of this procedure starts with the less expensive lactol 9 as chiral auxiliary [6]. In this case the ratio between the desired acetal 10 and the diastereomeric acetal 11 is favourably changed to 1,8 : 1. The higher stability against acids and the larger difference in the Rf-values of the two acetals led to a more convenient chromatographic separation. Again the undesired 11 and the small amounts of diacetal, which appeared as byproduct, were easily converted to 10 by acidic equilibration. 10 was transformed to the silylether 12 according to the above mentioned procedure. Due to its greater stability against acids a method had to be developed to hydrolyze the acetal in the presence of the silylether. Thus treatment with small amounts of methanolic hydrogen chloride in THF at elevated temperature led to the reusable methylated chiral auxiliary and high yields of 13, which was oxidized to enone 8 by Jones reagent.
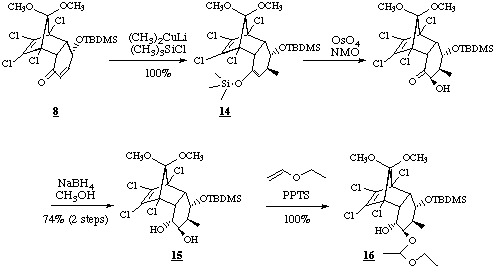
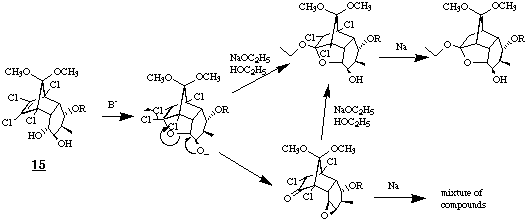
The steps to the decalinone 2 were performed according to the methods developed for the racemic decalinone (+/-)-2 [7]. Dimethyl copper lithium was added to the chiral enone 8 and the enolate formed was captured as silylenolether 14 in quantitative yield by addition of trimethylsilyl chloride and triethylamine. Within two steps this silylenol ether was transformed to the vicinal trans diol 15 by osmylation and reduction with sodium borohydride. Since intramolecular nucleophilic addition to the double bond with sodium ethanolate as base was pestered with side reactions due to the newly introduced unprotected hydroxy group in exo position, this function was protected as 1-ethoxyethylether.
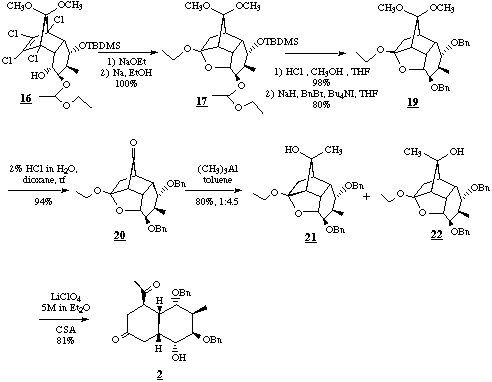
Treatment of 16 with sodium in refluxing ethanol allowed nucleophilic addition, substitution and dechlorination in one step in nearly quantitative yield. The protective groups of the tetracycle 17 were exchanged against the acid stable benzyl groups by methanolysis to the dihydroxy compound 18 and benzylation to 19. The dibenzyl derivative 19 was then hydrolysed to the monoketone 20, which was converted to the diastereomeric tertiary alcohols 21 and 22. Experiments with the racemic tertiary alcohols (+/-)-21 and (+/-)-22 had revealed that contrary to the model compounds [3c], acidic fragmentation occurred exclusively with alcohol (+/-)-22. After several trials trimethylalane in toluene at -78deg.C proved to be satisfactory in converting 20 to a 1 : 4.5 mixture of 21 and 22 in 80% yield. Treatment of 22 with small amounts of camphor sulfonic acid in 5M lithium perchlorate/ether solution at reflux did result in the desired chiral decalinone derivative 2 in 81% yield. Because of the drastic conditions of the last step the structure of 2 was confirmed by X-Ray diffraction analysis of a single crystal.
{kind=link}
{kind=link}