Key words: 3-thiazolines, 3-oxazolines, N-acylamino-4-oxomethyphosphonates of 3-thiazolidines and 3-oxazolidines
In the same manner, nomerous biological functionallities have been observed from the alkyloxyphosphonate esters. Amoung these are antiviral activity[4], pestiside[5], fungicide[6], inhibitors[7], insecticide[8], etc.
Scheme 1

Scheme 2
Considering the outstanding effects of 1-alkoxyphosphonates along with the biological activities of N-Acyl-4-alkoxy derivatives of 3-thiazolines and 3-oxazolines promoted us to synthesize the N-acylaminooxomethylphosphonates 4a-f.
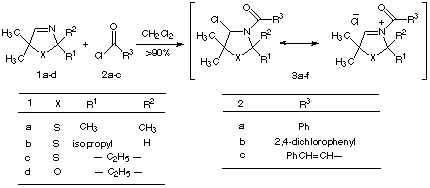
In this porpouse, the heterocyclic imines 1a-d were reacted with acid chlorides 2a-c. The resulting N-acyliminium salts 3a-f were then treated in the presence of triethylamine with 2-hydroxymethylphosphonic acid diethyl ester[9] to obtaine the N-acylaminooxomethylphosphonates 4a-f in crystalline low melting point to oil forms (Scheme 2). In the casese of 4b and 4c almost no distereoselectivity were observed. Table 1 outline the yields and the melting or boiling points of N-acylaminooxomethylphosphonates 4a-f.
Table 1
4 Yield[a] mp / bp a 48 48[b] b 38 28[b] c 40 130[c] d 50 160[c] e 50 45[b] f 35 140[c]
[a] Based on 1a-d. [b] mp in [[ring]]C , uncorrected. [c] The Kugelrohr distillation temperature at 0.01 mbar.
Experimental
Melting points were determined in an open capillary tube on a Dr. Linström instrument and were uncorrected. The elemental analyses were carried out on a Carlo Erba Stumentalione (MOD 1104) and the results were in agreement within satisfactory microanalysis +/- 0.4. The 1H-, 13C- and 31P-NMR spectra were recorded on a Bruker-Karlsruhe AM 300 spectrometer and chemical shifts were presented in ppm from the internal standards tetramethylsilane for 1H and 13C and phosphoric acid 85% for 31P measurements. The heterocyclic imines 1a-d1,2,10 as well 2-hydroxymethyl-phosphonic acid diethyl ester9 were prepared according to the literature. The mass spectroscopy data were recorded on a Finnigan-MAT 212(data system SS 300) spectrometer.
General procedure for the preparation of N-acylaminooxomethylphosphonates 4a-f: In a dry condition and under stirring, to a dichloromethane (15 mL) solution of heterocyclic imines 1a-d (10 mmol) , kept in 0[[ring]]C, was dropped a dichloromethane solution (10 mL) of acid chlorides 2a-c (10 mmol). The reaction mixture was left to stir at room temperature overnight and then cooled again to 0[[ring]]C. In 5 minutes interval, first, a solution of 2-hydroxymethyl-phosphonic acid diethyl ester10 (12 mmol) in absolute dichloromethane (10 mL), and then, triethylamine ( 5 mL) were dropped successively. The reaction mixture was stirred overnight at ambient temperature and then filltered off from the performed triethyl amine hydrochloride salt. The filtrate was washed with water (3x50 mL) and dried (MgSO4). The solvent was evaporated under reduced pressure and the unreacted 2-hydroxymethyl-phosphonic acid diethyl ester was completely distilled off in vaco (100[[ring]]C, 0.01 mbar). In the cases of 4a-b and 4e the colorless products were crystalized by dissolving the crude product in a mixture of diethyl ether (3 mL) and pethrolether (40/60, 3 mL) and standing overnight at -20[[ring]]C. Compounds 4c-d were obtained as colorless and light green oil, respectively, by Kugelrohr distillation in vaco. Compound 4f was obtained as colorless oil by subjecting the viscose crude product to flash chromatogeraphy on selica gel(eluent: diethyl ether/n-hexane, 5/1, Rf=0.38).
2,2,5,5-Tetramethyl-3-(3-phenyl-acryloyl)-thiazolidin-4-yloxymethyl-phosphoni acid diethyl ester (3a):
1H-NMR(CDCl3/δ): 1.08, 1.32(2t, 3J= 6.71 Hz, 6H, 2xCH3CH2); 1.48, 1.55, 2.02, 2.15(4s, 12H, 4xCH3); 3.74-3.96(m, 4H, 2xCH2O); 4.13-4.23(m, 2H, PCH2O); 5.82(s, 1H, C4-H); 7.13, 7.73(2d, 3J1= 15.26 Hz, 3J2= 15.23 Hz, 2H, CH=CH); 7.35-7.62(m, 5H, Ar-CH) ppm.
13C-NMR(CDCl3/δ): 16.20(m, 2xCH3CH2); 23.19, 30.99(4xCH3); 45.70(C5); 52.43, 58.30(2xCH2O); 62.24( JPC=34.7 Hz, PCH2O); 72.87(C2); 96.71(C4); 119.45, 144.10 (CH=CH); 127.90, 128.16, 128.73, 129.68, 129.96, 134.73(Ar.C); 165.80(C=O) ppm.
31P-NMR(CDCl3/δ): 19.84 ppm.
MS(CI, Isobutane) : m/e (%) = 442(90)[MH+].
C21H32NO5PS(441.2) Calc. C 57.12 H 7.31 N 3.17
found C 57.20 H 7.15 N 2.99
3-Bezoyl-2-isopropyl-5,5-dimethyl-thiazolidin4-yloxymethyl-phosphonic
acid diethyl ester (3b):1H-NMR(CDCl3/δ): 0.87, 0.98(2t, 3J=6.6 Hz, 6H, 2xCH3CH2); 1.21-1.32[m, 7H, CH(CH3)3]; 3.06-3.64(m, 4H, OCH2); 4.00-4.02, 4.14-4.21(m, 2H, OCH2P); 4.94, 5.12, (2s, broad, C4-H); 5.34, 5.52(2d, 3J=9.3 Hz, C2-H); 7.39-8.04(m, Ar-H) ppm.
13C-NMR(CDCl3/δ): 14.86, 15.00, 16.29, 16.36, 16.81, 20.18, 20.68, 22.41, 23.91, 27.50, 27.76, 30.89, 31.32, 31.44[6xCH3, CH); 51.59(C5); 54.97, 56.01, 58.25, 60,85(2xCH2O); 62.34-64.43(CH2OP); 68.65, 69.76(C2); 100.94(d, JPC=58.3 Hz, C4); 126.85-137.12(m, Ar-C); 171.00, 172.66(C=O) ppm.
31P-NMR(CDCl3/δ): 18.22, 18.55 ppm.
MS(CI, Isobutane) : m/e (%) = 430(100)[MH+].
C20H32NO5PS(429.2) Calc. C 55.92 H 7.51 N 3.26
found C 55.79 H 7.61 N 3.33
3-(2,4-Dichloro-benzoyl)-2-isopropyl-5,5-dimethyl-thiazolidin-4-yloxymethyl-posphonic acid diethyl ester (3c):
1H-NMR(CDCl3/δ): 0.87-0.98, 1,21-1.37, 1.57, 1.65, 1.72(m, 18 H, 6xCH3); 2.07-2.19 [m, 1H, CH(CH3)2]; 3.85-3.87, 4.11-4.32, 4.56-4.59(m, 10H, 3xCH2, C4-H); 5.37-5.40, 5.53-5.58(2m, 2x1H, C2-H); 7.26-7.83(m, 3H, Ar-H)ppm.
13C-NMR(CDCl3/δ): 14.75-29.32[m, 6xCH3, CH(CH3)2]; 32.76, 34.38(C-5); 53.99, 56.01, (2d, JPC-1= 37.1 Hz, JPC-2= 37.4 Hz, 2xCH2O); 58.49(m, OCH2P); 68.69(m. C4); 90.26, 91.34(C2); 116.81-135.93(m, ArC); 166.71, 168.32(C=O) ppm.
31P-NMR(CDCl3/δ): 17.51, 23.42 ppm.
MS(CI, Isobutane) : m/e (%) = 498(20)[MH+], 330(100)[MH++2].
C20H30Cl2NO5PS(497.1) Calc. C 48.28 H 6.08 N 2.82
found C 48.15 H 6.17 N 2.65
4-(2,2-Dichloro-benzoyl)-2,2-dimethyl-1-thia-4-aza-spiro[4.5]dec-3-yloxymethy) -phosphonic acid diethyl ester (3d):
1H-NMR(CDCl3/δ): 1.27(2t, 3J=6.80 Hz, 6H, 2xCH3CH2); 1.03-2.27(m, 16H, C5-CH3, cyclohexyl-CH2); 3.82-4.66(m, 7H, 3xCH2O, C4-H); 7.21-7.54(m, 3H, Ar-H) ppm.
13C-NMR(CDCl3/δ): 16.44, 24.14, 27.99, 30.15(4xCH3); 21.89-42.05(m, cyclohexyl-CH2); 52.23(C5); 56.36, 58.11(2xCH2O); 62.84(m, CH2OP); 80.21(C2); 90.92(d, JPC=56.4 Hz, C4); 127.10-132.74(m, Ar-C); 165.78(C=O) ppm.
31P-NMR(CDCl3/δ): 17.49 ppm.
MS(CI, Isobutane) : m/e (%) = 524(100)[MH+].
C22H32Cl2NO5PS(523.1) Calc. C 50.47 H 6.17 N 2.68
found C 50.33 H 6.28 N 2.55
4-Benzoyl-2,2-dimethyl-1-thia-4-aza-spiro[4.5]dec-3-yloxymethyl-phosphonic acid diethyl ester (3e):
1H-NMR(CDCl3/δ): 1.27(t, 3J=7.19 Hz, 6H, 2xCH3CH2); 1.38, 1.66(2s, 6H, 2xCH3); 1.21-2.17, 2.88-2.98, 3.12-3.29(m, 10H, cyclohexyl-CH2); 3.67-3.76, 4.00-4.11(2m, 6H, 3xCH2O); 5.20(s, 1H, C4-H); 7.41-7.49(m, 5H, Ar-CH) ppm.
13C-NMR(CDCl3/δ): 16.36(16.32, 16.39(2x CH3CH2); 23.30, 31.84(2xCH3); 24.35, 24.70, 25.82, 36.79, 38.13(cyclohexyl-CH2); 51.84(C5); 59.86, 62.11-62.60(3xCH2O); 79.89(C2); 100.21(d, JPC=55.2 Hz, C4); 127.54, 128.75, 130.27, 138.03(Ar-C); 170.80(C=O) ppm.
31P-NMR(CDCl3/δ): 18.90 ppm.
MS(CI, Isobutane) : m/e (%) = 456(45)[MH+].
C22H34NO5PS(455.2) Calc. C 58.00 H 7.53 N 3.08
found C 58.12 H 5.39 N 2.84
2,2-Dimethyl-4-(3-phenyl-acroyloyl)-1-oxa-4-aza-spiro[4.5]dec-3-yloxymethyl-posphonic acid diethyl ester MH359(3f):
1H-NMR(CDCl3/δ): 1.28(2t, 3J=6.80 Hz, 6H, 2xCH3CH2); 1.09-2.26(m, 16H, C5-CH3, cyclohexyl-CH2); 3.90-4.53(m, 7H, 3xCH2O, C4-H); 7.10-7.75(m, 7H, Ar-H, CH=CH) ppm.
13C-NMR(CDCl3/δ): 16.44, 24.14, 27.99, 30.15(4xCH3); 21.89-42.05(m, cyclohexyl-CH2); 52.23(C5); 56.36, 58.11(2xCH2O); 62.84(m, CH2OP); 80.21(C2); 90.92(d, JPC=56.4 Hz, C4); 127.10-132.74(m, Ar-C); 165.78(C=O) ppm.
31P-NMR(CDCl3/δ): 17.99 ppm.
MS(CI, Isobutane) : m/e (%) = 464(100)[MH+].
C24H34NO6P(463.2) Calc. C 62.17 H 7.40 N 3.02
found C 61.89 H 7.11 N 3.34
Acknowledegment: This research was supported, in part, by the Fonds der Chemischen Industrie, Deutsche Forschungsgemeinschaft and Degussa AG.
References: