Discussion
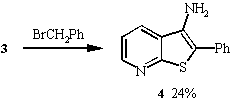
Treatment of 3 with benzyl bromide in the presence of sodium
ethoxide in DMF at room temperature for two hours gave 3-amino-2-phenylthieno[2,3-b]pyridine
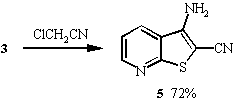
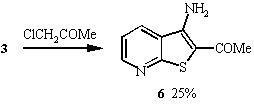
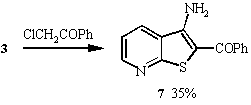
4 in 24% yield. Similarly, thienopyridines 5 -7
were prepared by reacting 3 with chloroacetonitrile, chloroacetone
and chloroacetophenone respectively under the same reaction conditions.
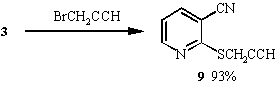
When the alkylating agents employed were halides containing unsaturated functionality, the products obtained were found to be pyridine derivatives 8, 9 and 10. Further treatment with base failed to convert these pyridines to the desired thieno[2,3-b]pyridines. Probably because these products do not have sufficiently acidic methylene protons to generate the desired anion for cyclisation. Oxidation of the sulphur to the sulphoxide might increase acidity and allow cyclisation.
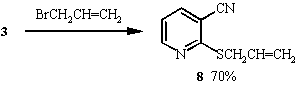
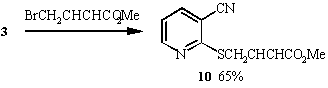
As well as haloalkanes and allylic halides,
a-haloesters were also employed as
alkylating agents. For example, 3-amino-2-carboethoxythieno[2,3-b]pyridine
11 was produced in 56% yield (when the alkylating agent
was ethyl chloroacetate). Its spectral data and melting point
were analogous to those previously reported8. It was
expected that the reaction of 3 with diethyl bromomalonate
would lead to the formation of 13 via diester 12.
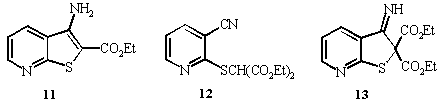
However, examination of the ir and nmr spectra for the product showed it
to be thienopyridine 11 (42% yield) formed by the
mechanism described. The reaction
proceeds as usual with initial base attack on the thione resulting
in alkylation of the sulfur atom. The CH proton is very
acidic and therefore is removed by base which promotes cyclisation possibly
to the thienopyridine 13. Regeneration of the base results
in attack on one of the ester carbonyl groups leading to de-esterification
and formation of 11 with loss of diethyl carbonate. [These
Claisen-type condensations9-14 readily occur with compounds
in which intramolecular condensation is possible.]
This prompted us to attempt the preparation of 6 in a similar manner by reaction of 3 with ethyl 2-chloroacetoacetate. The product obtained from this reaction (63%) also proved to be the ubiquitous thienopyridine 11. Surprisingly in this case the ketone was removed by ethoxide as ethyl acetate rather than loss of the ester group as diethyl carbonate. This was unexpected as it was believed that the carbon of the ester group might be more susceptable to nucleophilic attack since it contains an alkoxy group as well as a carbonyl group. However the fact that the ketone group was displaced may be because ethyl acetate is a better leaving group than diethyl carbonate. We propose a mechanism for this reaction.
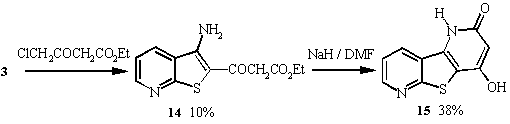
When 3 was treated with ethyl 4-chloroacetoacetate, two
products were isolated 14 and 15 in 10% and 38%
yields respectively (trituration with ethyl acetate solublised
14 but not 15). The structures were elucidated
by examination of the spectral data. The proposed mechanism proceeds
as usual with base attack on the thione resulting in alkylation
of the sulfur atom. The hydrogens of the CH2 group a
to the sulfur are extremely acidic and removal of a proton is
easily achieved with base. Cyclisation onto the nitrile forms
an imino intermediate which is tautomerised to the amine 14.
Intramolecular displacement of the ethoxide forms tricycle 15
after enolisation of the acidic CH2 proton.