 ECHET96 Article 109 Michael A. Walters
ECHET96 Article 109 Michael A. Walters
Solid-phase parallel synthesis applied to lead development:
potent analogues of the GPIIb/IIIa antagonist RWJ-50042
William J. Hoekstra,* Bruce E. Maryanoff, Patricia Andrade-Gordon, Judith H.
Cohen, Michael J. Costanzo, Bruce P. Damiano, Robert Falotico, Barbara J.
Haertlein, Bruce D. Harris, Jack A. Kauffman,
Patricia M. Keane, David F. McComsey, John A. Mitchell, Frank J. Villani, Jr.
and Stephen C. Yabut
Drug Discovery and Chemical Development Department,
The R. W. Johnson Pharmaceutical Research Institute, Spring House,
Pennsylvania 19477, USA
Abstract
A series of b-turn peptide mimetics with a nipecotic acid
heterocyclic scaffold was designed by NMR analysis of the C-terminal
g-chain of fibrinogen to provide the lead GPIIb/IIIa (fibrinogen receptor)
antagonist RWJ-50042 (1). We have employed solid-phase parallel
synthesis for the preparation of over 200 analogues of this lead with a
protocol of optimization cycles. This strategy produced some nipecotamide
analogues with a 100-fold improvement in potency.
Combinatorial chemistry methods have unleashed a powerful strategy for
diversity-based discovery of new leads.1 Convergent, resin-based
parallel synthesis of discrete chemical libraries is a focused adaptation of
combinatorial chemistry that provides an attractive lead development tool for
the refinement of biological activity.2 Herein, we report on the
solid-phase parallel synthesis of analogues of a prototypical fibrinogen
receptor (GPIIb/IIIa) antagonist lead, RWJ-50042 (1). This approach
resulted in a marked compression of the drug lead-to-clinical-candidate
timeline vis-a-vis traditional solution-phase techniques.
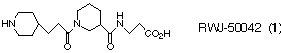
RWJ-50042 is an orally active antagonist of the platelet fibrinogen receptor
(binding IC50 = 0.0045 uM; sustained ex vivo inhibition of collagen-induced
platelet aggregation at 10 mg kg-1 in dogs), which was discovered by using the
solution structure of the C-terminal g-chain of fibrinogen as a basis for
drug design.3-7 This substituted nipecotic acid modelled the
b-turn structure contained within the KQADG sequence of the g-chain
(residues 406-410). Given the competitive environment surrounding this type of
antithrombotic therapy,8 we pursued synthetic methodology to
expedite the study of RWJ-50042 analogues.
Synthesis of analogues
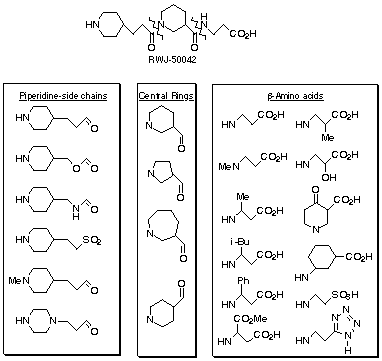
Solid-phase parallel synthesis was employed to prepare analogues of RWJ-50042
quickly. Given that this lead molecule consists of two amide
bonds/three structural components, variation of each of these elements
represents a practical, systematic strategy toward potency improvement (Scheme
1). Since variants of the b-amino acid component are readily available as
esters, commercially or synthetically, a dozen were selected initially for
fibrinogen receptor antagonist synthesis. Our apparatus is arranged in a
three-by-four array to effect production of twelve products for any given set
of molecules; the N-terminal pseudodipeptide unit is held constant in a matrix.
Importantly, we chose a strategy of N-terminal attachment to the resin to allow
for coupling of the numerous b-amino esters relatively late in the
synthesis, thereby timing the 12-way resin division immediately before the
second amide bond coupling (Scheme 2). A corresponding C-terminal attachment
strategy would have incorporated less-available N-protected b-amino acids
and compelled resin splitting prior to the first amide bond-forming step.
Scheme 1 Strategy for preparation of a "virtual" library of 288
variants of RWJ-50042 (6 x 4 x 12)
Isonipecotic acid and five- and seven-membered-ring variants of
nipecotic acid were chosen to study scaffold size/conformation change of the
"central ring" in relation to the other two components. While 288 variants (6
x 4 x 12) were targeted in principle for synthesis, a concurrent refinement
process was implemented to select relatively optimal components for subsequent
analogue synthesis/evaluation. This process of using a "virtual" compound
library discards inferior components to avoid the unnecessary synthesis and
bioassay of weakly active agents.
Resin-based preparation of the 3-phenyl-3-aminopropionic acid analogue
(15) of RWJ-50042 typifies our strategy of convergent, high-throughput
synthesis. N-attachment of 2-chlorotrityl chloride resin to allyl
4-piperidinepropanoate furnished "N-protected" intermediate A. Allyl
ester removal under mild, reproducible palladium(0) conditions and then
DIC-mediated coupling to allyl piperidinepropanoate afforded pseudo-dipeptide
B. Starting at the N-terminus allows one to accrue large quantities of
resin-bound intermediate B in a common reaction vessel. Once B
was saponified, the resin was split for coupling with twelve readily available
b-amino esters, leading to final products. For modifications at the
tertiary amide, analogous urethane and urea couplings at the N-nipecotyl
position were performed by using standard p-nitrophenylchloroformate
conditions9 with the appropriate resin-bound primary alcohol or
primary amine (e.g., 2 and 3). Solution-phase coupling of
CBZ-4-piperidineethanesulfonyl chloride with ethyl nipecotate gave the
corresponding sulfonamide intermediate (e.g., 4).10
Intermediate B was saponified and coupled to methyl
3-phenyl-3-aminopropionate to render C. To isolate a variety of
carboxylic acid targets from readily available methyl or ethyl b-amino
esters, an organic solution method of KOSiMe3/THF saponification was adapted to
intermediates such as C. This method allows the resin to swell suitably
to complete ester cleavage when basic aqueous conditions fail. Potassium
carboxylates were then acidified with dilute AcOH and cleaved with CF3CO2H to
give products, exemplified by 3-phenyl derivative 15.
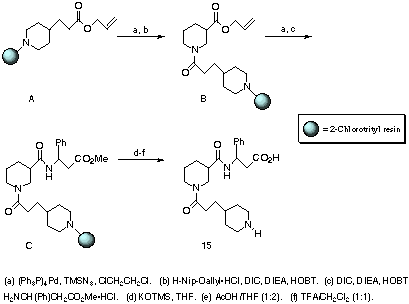
Scheme 2
Racemic 3-substituted b-amino esters were purchased, or prepared by using
a modified Knoevenagel procedure (RCHO/ammonium acetate/malonic
acid).11 Biological testing of racemic, matrix-produced targets
revealed highly active antagonists of interest for enantiospecific
solution-phase scale-up synthesis. Enantiomerically enriched 3-aryl-3-amino
esters were synthesized by using (R)-1-phenylethylamine Michael addition
to arylacrylates.12 Lithium acetylide addition to
4-benzoyloxy-2-azetidinone followed by ring opening and chromatographic chiral
resolution of the desired O-methylmandelamide derivative expedited
preparation of acetylene intermediates.13 Racemic
3-aryl-3-aminopropanoic acids were also resolved as their phenylacetamide
derivatives by using penicillin amidase (Scheme 3).14
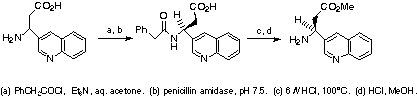
Scheme 3
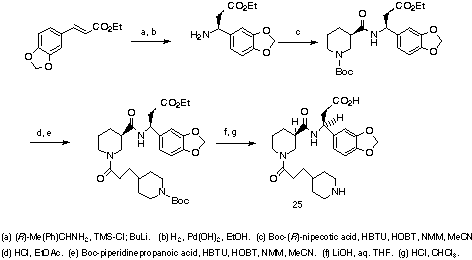
Scheme 4
A representative synthesis that uses the asymmetric Michael addition as
the key step is shown for methylenedioxybenzene derivative 25 (Scheme
4). Conjugate addition of (R)-(+)-1-phenylethylamine to ethyl
3,4-methylenedioxybenzeneacrylate proceeds with >90% diastereomeric excess. Hydrogenolysis of
the a-methylbenzyl group and HBTU-mediated coupling with
Boc-(R)-(-)-nipecotic acid15 gives a Boc-nipecotamide
intermediate which is then N-terminally deprotected and coupled iteratively
with Boc-piperidinepropanoic acid. Purifications were performed on fully
protected coupling products via chromatography on silica gel. Nipecotamide
25, for instance, was isolated in 17% overall yield.
Discussion
Compounds 1-25 are representative examples of more than 250 solid-phase
synthesis products that were tested in vitro (Table 1). Systematic changes at
the piperidine linker unit Y indicate that a tertiary amide at this site is
preferred (e.g., 1). While 12 b-amino acid variants of urethane
(2) or urea (3) linkers were prepared on the resin, sulfonamide
(4), N-Me-piperidine (5), and piperazine (6)
examples represent singular samplings from solution-phase synthesis. Since the
preferred central ring turned out to be nipecotic acid (n = 1, 1), about
180 variants were prepared while 12 each of examples 7-9 were isolated
and tested. In vitro testing of b-amino acid variants 12-22
indicated the possibility of activity improvement with 3-alkyl (13,
14), 3-aryl (15), or 2-oxy (18) substitutions. Aspartate
methyl ester 16 is regarded as a "3-alkyl" case since its diacid
derivative is inactive (IC50 > 25 uM). Testing of 29 alkyl, alkenyl, or
alkynyl 3-substituted compounds gave antagonists with as much as fourfold
potency improvement (e.g., phenylacetylene 23 and
tert-butylacetylene 24, Table 2) over the active enantiomer of
RWJ-50042 (i.e., 10). Large hydrophobic groups in this part of the
molecule rendered the best activity; thus, numerous 3-aryl cases were prepared
(120 compounds tested).
This parallel synthesis methodology rapidly produced greater than a dozen
analogues of RWJ-50042, which were targeted for enantiospecific scale-up
synthesis and subsequent in vivo evaluation (some are shown in Table 2). From
an in vitro standpoint, some of the more promising compounds were the doubly
racemic 3-(3,4-methylenedioxyphenyl) and 3-(3-quinoline) congeners. Indeed,
enantiospecific synthesis of these fibrinogen receptor antagonists afforded
the most potent compounds in this series (3S-enantiomers 25 and
28). 3-Substituted b-aminopropionic acid analogues with the
R absolute configuration are only weakly active.
To address potential oral bioavailability limitations of our amino acid-like
antagonists, compound 25 was "capped" at the N- or C-terminus.
N-Methylpiperidine 26 and ethyl ester 27 exhibit inferior
in vitro and in vivo characteristics relative to 25, however. The power
of parallel synthesis played a decisive role in overcoming this bioavailability
hurdle. Due to the ample selection of potent, solid-phase synthesis-derived
compounds, analogues were identified with useful systemic availability (15-20%)
without the need for prodrug modifications (oral canine studies).
Table 1 Inhibition of human platelet aggregation and fibrinogen
binding by RWJ-50042 analogues (uM)
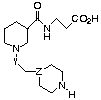
Pl. Agg. Bndg Pl. Aggr. Bndg
# Y Z IC50* IC50+ # n IC50*
IC50+
1 COCH2 CH 0.66 0.005 1 1 0.66 0.005
2 COO CH 4.7 0.027 7 0 7.0 0.004
3 CONH CH 6.7 0.016 8 2 7.6 0.013
4 SO2CH2 CH 11.0 0.025 9 1 >25 (isonipec.) >25
5 COCH2 CH 2.4 0.006 10 1 0.34 (3R) 0.005
(N-Me-piperidine) 11 1 2.93 (3S) 0.004
6 COCH2 N >25 >25
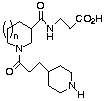
Pl. Agg. Bndg Pl. Agg. Bndg
# X IC50* IC50+ # X IC50*
IC50+
1 b-Ala 0.66 0.005 17 2-Me-b-Ala >25 >25
12 N-Me-b-Ala 27.5 0.20 18 2-OH-b-Ala 0.85
0.005
13 3-Me-b-Ala 2.0 0.003 19 4-oxo-nipecotic acid >25
0.33
14 3-Bui-b-Ala 4.1 0.0025 20 3-NH-c-C6H10-CO2H
>25 0.20
15 3-Ph-b-Ala 3.6 0.003 21 NH(CH2)2SO3H 10.8 0.18
16 L-Asp-OMe 1.1 0.003 22 NH(CH2)2-5-tetrazole 25.5
1.39
* Thrombin-induced gel-filtered platelet aggregation (uM, n =
3).3
+ Inhibition of biotinylated fibrinogen binding to immobilized
GPIIb/IIIa (uM, n = 2).3
Table 2 In vitro data for three-substituted b-amino acid
GPIIb/IIIa antagonists
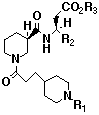
Fg Binding+ Human GFP*
# R1 R2 R3 IC50 (uM) IC50 (uM)
10 H H H 0.005 0.34
23 H C
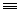 C-Ph
H 0.0002 0.080
C-Ph
H 0.0002 0.080
24 H C
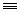 C-But
H 0.021 0.079
C-But
H 0.021 0.079
25 H 3,4-methylenedioxy-Ph H 0.0005 0.028
26 Me 3,4-methylenedioxy-Ph H 0.0002 0.84
27 H 3,4-methylenedioxy-Ph Et 0.046 36
28 H 3-quinoline H 0.0002 0.019
29 H 2-thiophene H 0.0002 0.090
Xemlofiban (SC-54684) 0.0006 0.31
* Thrombin-induced gel-filtered platelet aggregation (n = 3).3
+ Inhibition of biotinylated fibrinogen binding to immobilized
GPIIb/IIIa (n = 2).3
Table 3 Canine ex vivo data for four GPIIb/IIIa antagonists
Dog PRP* Dog ex vivo platelet
aggregation+
Compd IC50 (uM) PO Dose Duration
10 0.41 3 mg kg-1 120 min
24 0.45 10 mg kg-1 90 min
25 0.015 3 mg kg-1 >180 min
Xemlofiban 1.20 3 mg kg-1 >180 min
* Collagen-induced platelet-rich plasma aggregation (n = 3).
+ Oral dose required to inhibit collagen-induced canine ex vivo
platelet aggregation at least 50% (3 dogs).
Conclusion
Solid-phase parallel synthesis facilitated lead improvement/development of the
GPIIb/IIIa antagonists in the following manner. First, it produced numerous
potent analogues, in vitro and in vivo, of RWJ-50042 quickly.
Classical solution-phase synthesis would have, in all probability, resulted in a
similar quality of improvement, but not in the time period exhibited here.
Second, the number of improved antagonists yielded by resin-based synthesis
allowed for study of other critical in vivo properties such as consistency of
oral absorption, plasma halflife, duration of action, etc. Overall, the
timeline for lead discovery-to-development candidate selection within the
series was compressed relative to a traditional medicinal chemistry
approach.
Acknowledgment
We thank Dr Michael N. Greco for advice on the
solid-phase synthesis.
- (a) Gordon, E. M.; Barrett, R. W.; Dower, W. J.; Fodor, S. P. A.;
Gallop, M. A. J. Med. Chem. 1994, 37, 1385; (b) Thompson,
L. A.; Ellman, J. A. Chem. Rev. 1996, 96, 555.
-
DeWitt, S. H.; Kiely, J. S.; Stankovic, C. J.; Schroeder, M. C.; Cody, D. M.
R.; Pavia, M. R. Proc. Natl. Acad. Sci. USA 1993, 90,
6909.
-
Hoekstra, W. J.; Beavers, M. P.; Andrade-Gordon, P.; Evangelisto, M. F.;
Keane, P. M.; Press, J. B.; Tomko, K. A.; Fan, F.; Kloczewiak, M.; Mayo, K. H.;
Durkin, K. A.; Liotta, D. C. J. Med. Chem. 1995, 38,
1582.
-
Fan, F.; Mayo, K. H. J. Biol. Chem. 1995, 270, 24693.
-
Mayo, K. H.; Fan, F.; Beavers, M. P.; Eckardt, A.; Keane, P.; Hoekstra, W.
J.; Andrade-Gordon, P. FEBS Lett. 1996, 378, 79.
-
Mayo, K. H.; Fan, F.; Beavers, M. P.; Eckardt, A.; Keane, P.; Hoekstra, W.
J.; Andrade-Gordon, P. Biochemistry 1996, 35, 4434.
-
Hoekstra, W. J.; Press, J. B.; Bonner, M. P.; Andrade-Gordon, P.; Keane, P.
M.; Durkin, K. A.; Liotta, D. C. Bioorg. Med. Chem. Lett. 1994,
4, 1361.
-
For a recent review, see: Zablocki, J. A.; Rao, S. N.; Baron, D. A.; Flynn,
D. L.; Nicholson, N. S.; Feigen, L. P. Curr. Pharm. Design 1995,
1, 533.
-
Hutchins, S. M.; Chapman, K. T. Tetrahedron Lett. 1994,
35, 4055.
-
(a) DeGaw, J. I.; Kennedy, J. G. J. Heterocycl. Chem. 1966,
3, 90; (b) Hearst, P. J.; Noller, C. R. Org. Synthesis
1950, 30, 58.
-
Profft, E.; Becker, F. J. J. Prakt. Chem. 1965, 30,
18.
-
Rico, J. G.; Lindmark, R. J.; Rogers, T. E.; Bovy, P. R. J. Org.
Chem. 1993, 58, 7948.
-
Zablocki, J. A.; Rico, J. G.; Garland, R. B.; Rogers, T. E.; Williams, K.;
Schretzmann, L. A.; Rao, S. A.; Bovy, P. R.; Tjoeng, F. S.; Lindmark, R. J.;
Toth, M. V.; Zupec, M. E.; McMackins, D. E.; Adams, S. P.; Miyano, M.; Markos,
C. S.; Milton, M. N.; Paulson, S.; Herin, M.; Jacqmin, P.; Nicholson, N. S.;
Panzer-Knodle, S. G.; Haas, N. F.; Page, J. D.; Szalony, J. A.; Taite, B. B.;
Salyers, A. K.; King, L. W.; Campion, J. G.; Feigen, L. P. J. Med. Chem.
1995, 38, 2378.
-
Soloshonok, V. A.; Fokina, N. A.; Rybakova, A. V.; Shishkina, I. P.;
Galushko, S. V.; Sorochinsky, A. E.; Kukhar, V. P.; Savchenko, M. V.; Svedas,
V. K. Tetrahedron: Asymmetry 1995, 6, 1601.
-
Although (R)-(-)-nipecotamides were identified early on as more
potent antagonists than their epimers,3 racemic allyl nipecotate was
employed in high-throughput matrix synthesis. Boc-(R)-(-)-nipecotic
acid was prepared in two steps from (R)-(-)-nipecotic acid ethyl ester
(Chemi S.p.A.).